
Abstract
Background
Propionic acidemia (PA) is a serious metabolic disorder, and different approaches have been applied to its prenatal diagnosis. To evaluate the reliability and validity of a biochemical strategy in the prenatal diagnosis of PA, we conducted a retrospective study of our 11-year experiences at a single center.
Methods
We accumulated data from 78 pregnancies from 58 families referred to our center and provided prenatal diagnosis by directed genetic analysis and/or metabolite measurement using tandem mass spectrometry (MS/MS) and gas chromatography/mass spectrometry (GC/MS) of amniotic fluid (AF) samples.
Results
Sixty-five unaffected fetuses (83.33%) and 13 affected fetuses (16.67%) were confirmed in our study. The characteristic metabolites including propionylcarnitine (C3) level, C3/acetylcarnitine (C2) ratio and 2-methylcitric acid (2MCA) level in unaffected and affected groups showed significant differences (P < 0.0001), while the level of 3-hydroxypropionic acid (3HPA) showed no significant difference between the two groups (P > 0.05).Of the 78 pregnancies, 24 fetuses were found to have either one causative pathogenic variant or were without genetic information in the proband. Three of these fetuses had elevated AF levels of C3, C3/C2 ratio, and 2MCA and, thus, were determined to be affected, while the remaining fetuses were determined to be unaffected based on a normal AF metabolite profile. Our genetic and biochemical results were highly consistent with postnatal follow-up results on all unaffected fetuses.
Conclusions
We conclude that a biochemical approach can serve as a fast and convenient prenatal diagnostic method for pregnancies at an increased risk for PA, which could be used in conjunction with genetic testing for precise prenatal diagnosis of this disorder. In our analysis, the characteristic metabolites C3 level, C3/C2 ratio, and 2MCA level in AF supernatant were dependable biochemical markers for diagnosis, of which the C3/C2 ratio appears to be the most reliable biochemical marker for the prenatal diagnosis of PA.
Similar content being viewed by others


Introduction
Propionic acidemia (PA, OMIM #606054) is an organic acidemia attributed to the deficiency of propionyl-CoA carboxylase (PCC, EC6.4.1.3). PCC, which catalyzes the carboxylation of propionyl-CoA to D-methylmalonyl-CoA [1, 2], is a 750 kDa heterododecamer composed of 6 propionyl-CoA carboxylase alpha (PCCA) and 6 propionyl-CoA carboxylase beta subunits (PCCB) [3, 4]. Propionyl-CoA has a broad influence on metabolism, including impacts on the urea cycle, the citric acid cycle, and the glycine cleavage system [5]. PA has an autosomal recessive mode of inheritance and results from bi-allelic variants in PCCA or PCCB that impair enzyme function [6].
Although there are mild, late-onset forms of PA, most patients have a neonatal presentation with life-threatening metabolic decompensations [1, 7, 8]. Reliable methods for prenatal diagnosis are thus essential for neonatal management of PA. Different approaches have been applied to prenatal diagnosis of PA including direct propionyl-CoA carboxylase activity assay in chorionic villi (CV) [9, 10], quantification of the characteristic metabolites including acylcarnitines, propionic acids and methylcitric acids in cell-free amniotic fluid (AF) [11,12,13,14]; and direct pathogenic variant analysis of PCCA or PCCB genes in amniocytes. However, each approach has its limitations. Enzymatic assay can be time-consuming or unavailable, and activity can be influenced by the quality of the CV sample. Direct genetic analysis of amniocytes is dependent upon the availability of known familial mutations and diagnostic metabolites for PA in the AF can degrade over 1–2 weeks. Therefore, the use of two or more methods simultaneously may be needed to achieve the definitive prenatal diagnosis [15].
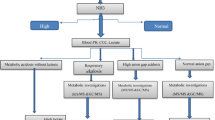
In the present study, we provide a retrospective review of our experience with prenatal diagnosis of PA over the last 11 years. Overall, we have investigated 78 pregnancies in 58 unrelated families by molecular genetics analysis of amniocytes and/or metabolite analysis in AF supernatants.
Materials and methods
Families and probands
In this study, 78 pregnancies (58 families) in which the probands diagnosed with PA were referred to our center seeking prenatal diagnosis from April 2008 to December 2019. The probands were diagnosed based on clinical symptoms, biochemical results and genetic testing of the PCCA or PCCB genes. Biochemical results included elevated blood levels of propionylcarnitine (C3) and propionylcarnitine to acetylcarnitine ratio (C3/C2) and elevated urine levels of 3-hydroxypropionic acid (3HPA) and 2-methylcitric acid (2MCA). The mutation spectrum of the PCCA and PCCB genes in this study is shown in Tables 1 and 2 and Table S2 [16,17,18,19,20,21,22,23]. Written informed consent was obtained from all participants, and our study was approved by the Ethics Committee of **nhua Hospital.
Amniocyte samples
Thirty milliliters of AF were obtained from each pregnant woman by amniocentesis performed between 16 and 20 weeks of gestation. Ten mL was used for DNA extraction and the remaining supernatant from this aliquot was used for the metabolite analysis. Twenty mL of AF was cultured for karyotype analysis, with the cultured amniocytes also available as a back-up.
Metabolite analysis of amniotic fluid
The levels of C3, C2 and C3/C2 were measured by MS/MS (Applied Biosystems, API 2000) using 3 μL of AF supernatant and the levels automatically calculated based on the assigned values for internal standards using ChemoView v1.2 software [24]. The organic acids levels in 2 mL of uncultured AF supernatant samples were measured using GC-MS (QP2010, Shimadzu Limited, Kyoto, Japan) as described by Hasegawa et al. [25].
Gene variant analysis
DNA from AF was extracted with a QIAamp DNA Blood Mini Kit (Qiagen Inc., Valencia, CA). We then conducted polymerase chain reaction (PCR) amplification and direct Sanger sequencing. Reference sequences PCCA (NM_000282.3) and PCCB (NM_000532.3) were obtained from the NCBI GENEBANK.
Linkage analysis
We collected 3–4 mL of peripheral blood from all PA pedigree members to perform a linkage analysis and to exclude maternal-cell contamination. Six short tandem repeats (STR) were selected according to informative STR loci in our laboratory, and we subsequently conducted the linkage analysis (STR and primers are listed in Table S1).
Statistical analysis
Reference ranges for the four analytes evaluated in this study were determined by a nonparametric approach, identifying the 99.5th percentiles of the cumulative levels from unaffected fetuses as the upper reference limit (C3: < 5 μmol/L; C3/C2: < 0.3; Propionic acid: < 35 mmol/mol Cr; 2MCA: < 0.5 mmol/mol Cr). Standard scatter plots were generated for level of C3, C3/C2 ratio, 2MCA and 3HPA in AFs. Wilcoxon rank sum tests with exact P values were performed to compare the C3, C3/C2 ratio, 2MCA and 3HPA level in affected and unaffected groups. P < 0.05 was considered statistically significant. Statistical analysis was performed using Prism 8 (GraphPad Software Inc.).
Results
We summarized the genetic and biochemical results of 78 prenatal samples from 58 families in our study (Table 1, Table S2), including 65 unaffected (83.33%) and 13 affected fetuses (16.67%). We found no sample with maternal contamination and uncovered no symptoms of PA upon postnatal follow-up in any of the 65 unaffected fetuses. For the 13 affected fetuses, however, the parents chose to terminate their pregnancies.
Biochemical analyses of amniotic fluid metabolites
Of 78 fetuses, 13 fetuses were determined to be affected based on metabolite tests (Table 1). The median (range) level of C3, C3/C2 ratio and 2MCA in affected samples were 11.45 μmol/L (5.07–43.69), 1.00 (0.62–2.11) and 1.19 mmol/mol Cr (0.61–4.58), respectively (Fig. 1). All of the individual levels of these three analytes in 13 fetuses was higher than that of defined reference ranges for each metabolite. However, the 3HPA levels in all affected samples, ranging from 0 to 25.18 mmol/mol Cr which were in the normal range.

Scatter-plot showing the distribution of characteristic metabolite levels in affected and unaffected amniotic fluids. a The distribution of C3 levels in AFs between affected and unaffected samples; b the distribution of C3/C2 ratios in AFs between affected and unaffected samples; c the distribution of 2MCA levels in AFs between affected and unaffected samples; d the distribution of 3HPA in AFs between affected and unaffected samples. Horizontal lines, median values. P values were determined by the Wilcoxon rank sum test, *P < 0.05, ** P < 0.01, *** P < 0.001
Sixty-five AF supernatant samples were determined to be unaffected according to our metabolite test results (Table S2). The median (range) level of C3, C3/C2 ratio, and propionic acid, and 2MCA in 65 unaffected samples were 1.56 μmol/L (0.61–5.87), 0.14 (0.05–0.28), 2.31 mmol/mol Cr (0–80.65), and 0 mmol/mol Cr (0–0.52), respectively (Fig. 1). There were seven unaffected fetuses that had a slightly increased level of one of the four biochemical markers studied (Table 2). We noted two fetuses (F008 and F053) with slightly higher levels of C3, while the C3/C2 ratio and levels of 3HPA and 2MCA were in the normal range. Four fetuses (F014, F042, F043, and F044) were found to possess higher levels of propionic acid, but had a normal C3/C2 ratio and C3 and 2MCA levels. We found one fetus (F020) with a slightly higher level of 2MCA, but a normal C3/C2 ratio and normal levels of C3 and propionic acid.
Although the metabolite results for the C3, C3/C2 ratio, and 2MCA in unaffected and affected groups showed significant differences (P < 0.0001), the level of 3HPA in AF samples showed no significant difference between the two groups (P > 0.05, Fig. 1).
Genetic analysis of the pathogenic variants in amniocyte DNA
There were 54 pregnancies with clear information as to pathogenic variants in probands and parents, of which 19 pregnancies were from families affected by pathogenic variants of the PCCA gene, while the other 35 pregnancies were from families affected by pathogenic variation in the PCCB gene. Twelve fetuses carried no pathogenic variants in PCCA or PCCB (22.22%), 32 fetuses carried a heterozygous variants (59.26%), and 10 fetuses carried compound heterozygous variants (18.52%; Tables 1 and 2, Table S1). To explore the relationship between genotype and levels of metabolites in AF, we compared the median level of C3, C3/C2 ratio, and propionic acid and 2MCA in PCCA pathogenic variant carriers (n = 12) or PCCB pathogenic variant carriers (n = 20) with reference samples and observed that neither PCCA nor PCCB pathogenic variant carriers showed significant differences in these four metabolite levels relative to the reference group.
Genetic and metabolite analyses of the 54 fetuses showed high consistency. Ten of 54 fetuses have been determined to be affected fetuses by molecular genetics analysis (Table 1). Metabolite analyses in AF were consistent with these results. AF samples in the 10 affected fetuses had elevated C3 level, C3/C2 ratio, and 2MCA level and we therefore also inferred them to be affected fetuses by metabolite analysis (Table 1). Forty-three of 54 fetuses were determined to be unaffected by genetic testing, including 31 carriers and 12 fetuses without identifiable mutations with normal levels of C3, C3/C2 ratio and 2MCA. There was only one fetus with inconsistent molecular genetics and metabolite results (Table 2). The fetus (F053) with a pathogenic variant inherited from the father had an AF C3 level at the cutoff of (C3 5.00 μmol/L). However, the other diagnostic metabolic markers were within normal limits. Thus, the overall metabolite panel is indicative of an unaffected fetus, as supported by molecular testing.
There were 24 fetuses with incomplete molecular genetics results. Three of these cases (F001, F017 and F023) had elevated C3, C3/C2 ratio and 2MCA in the AF and were thus determined to be affected (Table 1). The other 21 fetuses were determined to be unaffected by a normal metabolite profile, which was confirmed by a normal phenotype at postnatal follow-up. Of the 21 unaffected fetuses by biochemical testing, there were three fetuses (F014, F043 and F044) that showed high levels of propionic acid, but normal levels of the key diagnostic markers, were determined to be unaffected (Table 2 and S2). While fetus F008 was found to have a slightly elevated C3 level, the other diagnostic markers were normal, thereby determined to be unaffected. Similarly, fetus F020 had a only slightly higher 2MCA level, but with normal C3 and C3/C2 levels, was deteremined to be unaffected. Postnatal follow-ups for all 24 fetuses determined to be unaffected by biochemical testing were consistent with unaffected phenotype.
Discussion
PA is a rare organic acidemia with an average estimated incidence of ~ 1:100,000–150,000 in the wordwide [26, 27]. The typical presentation of PA entails metabolic decompensations, cardiac complications, chronic kidney disease, or encephalopathy [28]. Prognosis of PA is generally poor and strongly influenced by the duration of coma and the level of blood ammonia [29, 30], and severe patients without proper treatment can die in the newborn period or later. Although a series of treatment strategies have been implemented over the past decade to improve the survival rate and life quality of PA patients [5, 31, 32], they remain at high risk for life-threatening complications. Prenatal diagnosis especially in families of probands with PA, is an essential strategy for neonatal planning. In the present study, we describe our experiences with prenatal diagnosis of PA in 78 pregnancies over 11 years.
The measurement of metabolite levels in AF and stable ranges across gestational ages can assist the prenatal diagnosis of PA [33,34,35]. The analyses of acylcarnitines by MS/MS and organic acids by GC/MS in AF allowed us to rapidly and reliably diagnose this condition, similar to our experience with prenatal diagnosis of methylmalonic acidemia [36]. Prenatal diagnosis of PA using biochemical methods required systematic evaluation. The majority of previously reported studies were based on single case or small series cases. We analyzed AF supernatants from 78 pregnancies at risk for PA using MS/MS and GS/MS techniques and showed that the measurements of three metabolite indices (C3, C3/C2 ratio and 2MCA) allowed for fast prenatal diagnosis of PA. Importantly, this metabolite panel required only a very small amount of AF supernatant, yet provided results for multiple metabolites. The propionylcarnitine (C3) level, C3/C2 ratio and 2-methylcitric acid (2MCA) level in unaffected and affected groups showed significant differences (P < 0.0001). 3HPA levels were also determined, but were found not to be informative, as levels in affected and unaffected cases showed no significant difference between the two groups (P > 0.05). The C3/C2 ratio in AFs showed no overlap between affected and unaffected fetuses, while C3, 2MCA and 3HPA levels overlapped. For 54 fetuses with confirmed pathogenic variants in the probands, the results for the C3/C2 ratio and 2MCA were consistent with the genetic results. In contrast, the levels of 3HPA in both groups showed discrepancies with the genetic results. These results suggested that the C3 level, C3/C2 ratio, and 2MCA level in AF samples were better suited than 3HPA as metabolic markers for the prenatal diagnosis of PA and that the C3/C2 ratio appeared to be the most reliable of the four tested biochemical markers. The biochemical testing approach could provide a strong support for diagnosing PA using amniotic fluid supernatant. And in the future, we could adjust cut-off value of the parameters by taking into account the molecular genetics testing to achieve a higher specificity.
Molecular genetics testing is a vital standard for the prenatal diagnosis of PA that requires the exclusion of maternal contamination to avoid false test results. However, due to the high genetic heterogeneity or novel variants whose clinical appearance is unknown, a genetic diagnosis of PA may lead to misdiagnosis [37], and the method depends upon availability of genetic information from the proband and parents. In some PA families, only one causative pathogenic variant was found in the proband, or a genetic testing was not performed, leading to the inability to make a precise prenatal diagnosis using genetic testing alone. For these families, metabolite testing could provide fast diagnosis and enable them to make an informed decision. In our study, there were 24 fetuses with incomplete genetic results in the probands: three samples were determined to be affected according to elevated levels of C3, C3/C2 ratio, and 2MCA in the AF supernatants. The remaining 21 fetuses were inferred to be unaffected fetuses based on normal metabolite levels. Additionally, postnatal follow-up showed a normal phenotype in all unaffected fetuses. Hence, biochemical testing appears to be a suitable option in diagnosis and provides further information that can be used for genetic counseling.
Conclusion
In summary, prenatal diagnosis of PA should be offered to families with an affected proband. Although a genetic approach is routine for the prenatal diagnosis of PA, a biochemical approach would offer an additional reliable method, especially for families with incomplete genetic results. A combined genetic and biochemical approach would comprise the strongest strategy, providing increased diagnostic accuracy.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- PA:
-
Propionic acidemia
- PCC:
-
Propionyl-CoA carboxylase
- PCCA :
-
6 propionyl-CoA carboxylase alpha subunits
- PCCB :
-
6 propionyl-CoA carboxylase beta subunits
- MS/MS:
-
Tandem mass spectrometry
- GC/MS:
-
Chromatography/mass spectrometry
- C3:
-
Propionylcarnitine
- C2:
-
Acetylcarnitine
- C3/C2:
-
Propionylcarnitine to acetylcarnitine ratio
- 2MCA:
-
2-methylcitric acid
- AF:
-
Amniotic fluid
- 3HPA:
-
3-hydroxypropionic acid
- CV:
-
Chorionic villi
References
Wongkittichote P, Ah Mew N, Chapman KA. Propionyl-CoA carboxylase - a review. Mol Genet Metab. 2017;122(4):145–52.
Lucke T, Perez-Cerda C, Baumgartner M, Fowler B, Sander S, Sasse M, et al. Propionic acidemia: unusual course with late onset and fatal outcome. Metabolism. 2004;53(6):809–10.
Lamhonwah AM, Barankiewicz TJ, Willard HF, Mahuran DJ, Quan F, Gravel RA. Isolation of cDNA clones coding for the alpha and beta chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc Natl Acad Sci U S A. 1986;83(13):4864–8.
Lau EP, Cochran BC, Munson L, Fall RR. Bovine kidney 3-methylcrotonyl-CoA and propionyl-CoA carboxylases: each enzyme contains nonidentical subunits. Proc Natl Acad Sci U S A. 1979;76(1):214–8.
Haijes HA, Hasselt PM, Jans JJM, Verhoeven-Duif NM. Pathophysiology of propionic and methylmalonic acidemias. Part 2: treatment strategies. J Inherited Metab Dis. 2019;42(5):745–61.
Gravel RA, Lam KF, Scully KJ, Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977;29(4):378–88.
Kolker S, Cazorla AG, Valayannopoulos V, Lund AM, Burlina AB, Sykut-Cegielska J, et al. Erratum to: the phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015;38(6):1155–6.
Jiang YZ, Shi Y, Shi Y, Gan LX, Kong YY, Zhu ZJ, et al. Methylmalonic and propionic acidemia among hospitalized pediatric patients: a nationwide report. Orphanet J Rare Dis. 2019;14(1):292.
Perez-Cerda C, Perez B, Merinero B, Desviat LR, Rodriguez-Pombo P, Ugarte M. Prenatal diagnosis of propionic acidemia. Prenat Diagn. 2004;24(12):962–4.
Chadefaux B, Augereau C, Rabier D, Rocchiccioli F, Boue J, Oury JF, et al. Prenatal diagnosis of propionic acidemia in chorionic villi by direct assay of propionyl CoA carboxylase. Prenat Diagn. 1988;8(2):161–4.
Van Hove JL, Chace DH, Kahler SG, Millington DS. Acylcarnitines in amniotic fluid: application to the prenatal diagnosis of propionic acidaemia. J Inherit Metab Dis. 1993;16(2):361–7.
Naylor G, Sweetman L, Nyhan WL, Hornbeck C, Griffiths J, Morch L, et al. Isotope dilution analysis of methylcitric acid in amniotic fluid for the prenatal diagnosis of propionic and methylmalonic acidemia. Clin Chim Acta. 1980;107(3):175–83.
Fensom AH, Benson PF, Chalmers RA, Tracey BM, Watson D, King GS, et al. Experience with prenatal diagnosis of propionic acidaemia and methylmalonic aciduria. J Inherit Metab Dis. 1984;7(Suppl 2):127–8.
Inoue Y, Ohse M, Shinka T, Kuhara T. Prenatal diagnosis of propionic acidemia by measuring methylcitric acid in dried amniotic fluid on filter paper using GC/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;870(2):160–3.
Dionisi-Vici C, Deodato F, Roschinger W, Rhead W, Wilcken B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis. 2006;29(2–3):383–9.
Hu YH, Han LS, Ye J, Qiu WJ, et al. Gene mutation analysis in patients with propionic acidemia. Zhonghua Er Ke Za Zhi. 2008;46(6):416–20.
Richard E, Desviat LR, Perez B, Perez-Cerda C, et al. Genetic heterogeneity in propionic acidemia patients with alpha-subunit defects. Identification of five novel mutations, one of them causing instability of the protein. Biochim Biophys Acta. 1999;1453(3):351–8.
Kim SN, Ryu KH, Lee EH, Kim JS, et al. Molecular analysis of PCCB gene in Korean patients with propionic acidemia. Mol Genet Metab. 2002;77(3):209–16.
Tahara T, Kraus JP, Ohura T, Rosenberg LE, et al. Three independent mutations in the same exon of the PCCB gene: differences between Caucasian and Japanese propionic acidaemia. J Inherit Metab Dis. 1993;16(2):353–60.
Chen Z, Wen P, Wang G, Hu Y, et al. Analysis of PCCA and PCCB gene mutations in patients with propionic acidemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015;32(1):26–30.
Campeau E, Dupuis L, Leon-Del-Rio A, Gravel R. Coding sequence mutations in the alpha subunit of propionyl-CoA carboxylase in patients with propionic acidemia. Mol Genet Metab. 1999;67(1):11–22.
Ugarte M, Perez-Cerda C, Rodriguez-Pombo P, Desviat LR, et al. Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat. 1999;14(4):275–82.
Campeau E, Desviat LR, Leclerc D, Wu X, et al. Structure of the PCCA gene and distribution of mutations causing propionic acidemia. Mol Genet Metab. 2001;74(1–2):238–47.
Han LS, Ye J, Qiu W, Gao X, Wang Y, et al. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis. 2007;30:507–14.
Hasegawa Y, Iga M, Kimura M, Shigematsu Y, Yamaguchi S. Prenatal diagnosis for organic acid disorders using two mass spectrometric methods, gas chromatography mass spectrometry and tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;823(1):13–7.
Gu X, Wang Z, Ye J, Han L, Qiu W. Newborn screening in China: phenylketonuria, congenital hypothyroidism and expanded screening. Ann Acad Med Singap. 2008;37(12 Suppl):107–4.
Almási T, Guey LT, Lukacs C, Csetneki K, Vokó Z, Zelei T. Systematic literature review and meta-analysis on the epidemiology of propionic acidemia. Orphanet J Rare Dis. 2019;14(1):40–8.
Shchelochkov OA, Manoli I, Sloan JL, Ferry S, Pass A, Van Ryzin C, et al. Chronic kidney disease in propionic acidemia. Genet Med. 2019;21(12):2830–5.
Chapman KA, Gropman A, MacLeod E, Stagni K, Summar ML, Ueda K, et al. Acute management of propionic acidemia. Mol Genet Metab. 2012;105(1):16–25.
Aldubayan SH, Rodan LH, Berry GT, Levy HL. Acute illness protocol for organic acidemias: methylmalonic acidemia and propionic acidemia. Pediatr Emerg Care. 2017;33(2):142–6.
Nashabat M, Obaid A, Al Mutairi F, Saleh M, Elamin M, Ahmed H, et al. Evaluation of long-term effectiveness of the use of carglumic acid in patients with propionic acidemia (PA) or methylmalonic acidemia (MMA): study protocol for a randomized controlled trial. BMC Pediatr. 2019;19(1):195.
Chu T-H, Chien Y-H, Lin H-Y, Liao H-C, Ho H-J, Lai C-J, et al. Methylmalonic acidemia/propionic acidemia – the biochemical presentation and comparing the outcome between liver transplantation versus non-liver transplantation groups. Orphanet J Rare Dis. 2019;14(1):73.
Sweetman L, Weyler W, Shafai T, Young PE, Nyhan WLJJ. Prenatal diagnosis of propionic acidemia. J Am Med Assoc. 1979;242(10):1048–52.
Inoue Y, Kuhara T. Rapid and sensitive method for prenatal diagnosis of propionic acidemia using stable isotope dilution gas chromatography-mass spectrometry and urease pretreatment. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776(1):71–7.
Ottolenghi C, Abermil N, Lescoat A, Aupetit J, Beaugendre O, Morichon-Delvallez N, et al. Gestational age-related reference values for amniotic fluid organic acids. Prenat Diagn. 2010;30(1):43–8.
Ji X, Wang H, Ye J, Qiu W, Zhang H, Liang L, et al. Prenatal diagnosis of methylmalonic aciduria from amniotic fluid using genetic and biochemical approaches. Prenatal Diagn. 2019;39(11):993–7.
Wang H, Meng L, Li W, Du J, Tan Y, Gong F, et al. Combinations of exonic deletions and rare mutations lead to misdiagnosis of propionic acidemia. Clin Chim Acta. 2020;502:153–8.
Acknowledgements
We acknowledge all patients and their family for participating in this study.
Funding
This work was supported by The National Key Research and Development Program of China (No. 2016YFC0901505); Institute of Shanghai Municipal Commission of Health and Family Planning (Grant No.20174Y0219); and the National Natural science Foundation of China (Grant No. 81701458).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. LSH: study concept and design. MYD and BX: acquisition and analysis of data; drafting of the manuscript. ZX, WJX: performed MS/MS and GS/MS; of AFs; YW: performed genetic testing of AFs; LSH, BX, HWZ, J Y, WJQ, HZ, LW, LLL, GXF and YYG: study supervision; in charge of the patients after referral to each center and further diagnosis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all participants, and our study was approved by the Ethics Committee of **nhua Hospital (Approval No. XHEC-D-2020-028).
Consent for publication
Not applicable.
Competing interests
All authors declare no conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
STR sites and primers used for excluding maternal cell contamination. Table S2. The prenatal results of genetic tests and biochemical analysis in the amniotic fluid samples of 65 unaffected fetuses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dai, M., **ao, B., Zhang, H. et al. Biochemical and genetic approaches to the prenatal diagnosis of propionic acidemia in 78 pregnancies. Orphanet J Rare Dis 15, 276 (2020). https://doi.org/10.1186/s13023-020-01539-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01539-w