
Abstract
Background
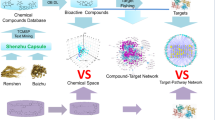
Pulsatilla decoction (Bai-Tou-Weng-Tang, BTWT) is a classic formula prescription of a traditional Chinese medicine that is used to treat ulcerative colitis (UC). However, its active components and underlying mechanism of action remain unclear. In the present study, we aimed to identify potential immunomodulators from BTWT that act at therapeutic targets for UC.
Methods
The protective effects of BTWT granules were examined in mice with colitis induced by dextran sulfate sodium. The absorbed components of BTWT were identified using LC-MS, and selected protein targets of these components in UC were investigated using molecular docking.
Results
Oral administration of BTWT granules significantly alleviated disease severity and colon shortening, and inhibited the inflammatory response in mice with chronic colitis. In these mice, 11 compounds from the BTWT granules were detected in the serum and/or colon. The molecular docking study demonstrated that compounds from Radix pulsatillae, such as anemoside A3, interacted with STAT3 and S1PR1; compounds from Rhizoma coptidis and/or Cortex phellodendri, such as palmatine, interacted with JAK3, PD-1, and PD-L1; and components of Cortex fraxini such as aesculin interacted with S1PR1, JAK3, STAT3 and PD-L1. Further in-vitro experiments showing that the compounds inhibited TNF-α and IL-6 production and STAT3 activation in RAW 264.7 cells suggested that these compounds have immunomodulatory activities.
Conclusion
We revealed for the first time that 11 absorbed ingredients from BTWT were immunomodulators against therapeutic targets for UC. These findings suggest that the identified compounds are the active components of BTWT, and the identified protein targets underlie the mechanism of action of BTWT against UC.
Similar content being viewed by others

Introduction
Pulsatilla decoction, also named Bai-Tou-Weng-Tang (BTWT) in traditional Chinese medicine (TCM), has been used as a classic formula prescription in China for treating human diseases caused by bacteria for thousands of years [1]. The use of BTWT was first recorded in Zhang Zhong**g’s Treatise on Febrile Diseases, a famous classical book of TCM in China. In TCM theory, ulcerative colitis (UC) occurs mainly due to an excess of damp-heat, and BTWT functions by clearing away internal heat and detoxifying, cooling blood, and stop** bleeding. Therefore, BTWT is widely used for treating UC in TCM, and its efficacy has been verified by clinical observations and in experimental models of UC [Full size image
In the 18 days colitis model, mice exposed to three cycles of 2.0% DSS for 4 days had a significant increase in the DAI at all time points compared with water-treated control mice (Fig. 3E). Compared with the DSS-treated control, the therapeutic administration of 531.5 mg/kg BTWT from day 5 significantly reduced the DAI at the end of treatment (day 18). Furthermore, the colon shortening and swelling caused by three cycles of DSS challenge were significantly antagonized by BTWT treatment (Fig. 3F–H). Further, BTWT treatment significantly reduced the DSS challenge-associated increase in the mRNA levels of TNF-α, IL-6, and IL-17 in the colon of mice with chronic relapsing UC (Fig. 3I–K).
The absorbed constituents from orally administered BTWT granules
Individual ingredients known to constitute the raw materials of the BTWT were analyzed as standards using the same chromatogram conditions to confirm that the peaks obtained from the UPLC analysis represented recognized constituents of BTWT (Fig. 4). A total of 11 compounds identified by UPLC-Q/TOF–MS analysis were assigned to an original herbal medicine known to be present in BTWT (Fig. 1). To define the absorbed chemical constituents, mass spectrometry data of standard chemicals was compared with mass spectrometry data of the absorbed compounds distributed in the circulation and colon tissue of mice. As demonstrated in Fig. 4, three compounds, 23-HA, epiberberine, and aesculetin, were only detected in mice colon samples. The remaining components, namely, AB4, AA3, berberine, palmatine, coptisine, jateorhizine, phellodendrine, and aesculin, were detected in both colon and plasma samples (Fig. 4).


Representative UPLC–QQQ-MS chromatograms of the identified components from R. pulsatilla A, C. fraxini B, C. phellodendri, and R. coptidis C in mouse plasma and colon samples after oral administration of 531.5 mg/kg BTWT (n = 5 mice per treatment group)
Binding of BTWT constituents to JAK3
As shown in Additional file 1: Fig. S1A, tofacitinib formed two hydrogen bonds with JAK3; Leu905 acted as a H-acceptor for the amine atom of the pyrrole moiety, and Glu903 acted as a H-donor for the pyrimidine moiety with the lowest calculated binding energy conformation value of -5.93 kcal/mol. This result was similar to results from two previous studies [37, 38]. In addition, the pyrrole moiety formed a π-H interaction with Leu828, and pyrimidine moiety formed two π-H interactions with Leu828 and Leu956 of JAK3. When docked to JAK3, all the tested compounds of BTWT achieved the lowest calculated binding energy conformation values ranging from −12.90 to −4.30 kcal/mol. As shown in Fig. 5A, only one component of R. pulsatilla, AA3, interacted with JAK3 in a way similar to that of tofacitinib; AA3 formed a hydrogen bond with Leu905; and Leu905 acted as an H-acceptor interacting with the oxygen atom of the ester group of AA3 (Fig. 5A). Notably, two components of C. fraxini formed numerous interactions with JAK3; these interactions also occur when tofacitinib is docked to JAK3. Specifically, the phenyl ring and/or the pyranoid ring of benzopyran-2-one of aesculin, aesculetin formed π-H interactions with Leu828 and/or Leu956 of JAK3 (Fig. 5B). Also, the hydroxyl groups of aesculetin formed a hydrogen bond with Glu903 of JAK3; Glu903 acts as a H-donor, similar to what is seen with tofacitinib binding (Fig. 5B). By contrast, four components of R. coptidis and C. phellodendri, namely, berberine, palmatine, coptisine, and epiberberine, formed π–H interactions with Leu828 of JAK3 through their phenyl ring and the pyridine ring of the isoquinoline ring (Fig. 5C).

Molecular docking results of AA3 A, aesculin, aesculetin B, berberine, palmatine, coptisine, and epiberberine C in the binding sites of JAK3 with the lowest calculated binding energy conformations. In the 2D images, polar and nonpolar residues in the active site of JAK3 are shown in purple and green, respectively. Hydrogen bonds in the sidechain and backbone are shown in red and blue arrows, respectively; the arrows point to the H-acceptor. The π-H interactions are marked with a red line and an arene-H label. The metal contact interactions are shown with a purple line. In the 3D images, the carbon atoms of the compounds are shown in yellow, and other atoms (e.g., oxygen, nitrogen, and sulfur atoms, etc.) are shown in blue. The amino acid residues of the JAK3 proteins that interacted with the compounds are shown in green. The formation of hydrogen bonds and π-H and π-π interactions are shown by a red dotted line
Binding of BTWT constituents to STAT3
The SH2 domain of STAT3 is the main target of small-molecule inhibitors. Hence, the pocket-type subdomain structures of STAT3 including the pTys705 binding sites (Lys591, Arg609, Ser611, and Ser613) were selected as binding pockets [39]. As shown in Additiional file 1: Fig. S1A, with the lowest calculated binding energy conformation value of −7.45 kcal/mol, SD-36 formed four hydrogen bonds with STAT3; Arg609, Ser611, Glu612, and Ser613 acted as H-acceptors for the interaction with oxygen atoms of the phosphate moiety of SD-36. SD-36 also formed a hydrogen bond with STAT3, and Glu638 acted as an H-donor for the amine atom of the indole ring. Moreover, one of the oxygen atoms of the phosphate moiety also formed a metal contact with Arg609. All tested compounds present in BTWT were docked to STAT3 and obtained the lowest calculated binding energy conformation values ranging from −8.54 to −2.53 kcal/mol. AB4 had the lowest energetic conformation value (−8.54 kcal/mol), and this was the only value that was comparable to that of SD-36 (−7.54 kcal/mol). Two R. pulsatilla components, AA3 and 23-HA, and one C. fraxini component aesculin formed hydrogen bonds with STAT3, which were also observed with SD-36 binding (Fig. 5A). Specifically, the hydroxyl groups of AA3 formed three hydrogen bonds with STAT3, with Ser611 and Sre613 as H-acceptors and Glu638 as an H-donor. In addition, 23-HA formed hydrogen bonds with STAT3; Arg609 and Ser611 acted as the H-acceptor for the ionized oxygen atom of the ester group, and Glu612 and Ser613 acted as the H-acceptor for the oxygen atom of the ester group.
Binding of BTWT constituents to S1PR1
The lowest calculated binding energy conformation value of S1P docked to the active site of S1PR1 was -12.90 kcal/mol, which is lower than that of ML056 (W146) (-10.6 kcal/mol). S1P formed hydrogen bonds with Asn101 of S1PR1 acting as the H-donor for the protonated amine of the alkyl chain, and with Glu294 of S1PR1 acting as the H-donor for the protonated amine hydroxyl group; this binding mode was also seen by Yuan et al. [40]. In contrast, the phosphate moiety of ML056 (W146) formed hydrogen bonds with Try29 and Arg120 and formed a metal contact with Arg120 (Additional file 1: Fig.S1A); this binding mode has been reported by several studies [41,42,43]. Docking of components contained in BTWT to S1PR1 gave the lowest calculated binding energy conformation values, ranging from -13.00 to -5.84 kcal/mol; AA3 had the lowest value. As shown in Fig. 6B, the oxygen atom of the glucopyranosyl or hydroxyl group of AB4, AA3, and aesculin formed hydrogen bonds with S1PR1 with Lys34 acting as an H-acceptor. This binding mode was also observed in the interaction of the two positive-control ligands and S1PR1. Furthermore, the hydroxyl groups of AB4, AA3, and aesculin formed hydrogen bonds with Glu294 of S1PR1 acting as the H-donor. The above two binding modes were also observed in the S1P–S1PR1 interaction (Additional file 1: Fig.S1A). These observations indicate that AA3, aesculin, and especially AB4 are highly likely to be potent S1PR1 modulators.

Molecular docking results of AB4, AA3, 23-HA, and aesculin in the binding sites of STAT3 A and S1PR1 B with the lowest calculated binding energy conformations. In the 2D images, the polar and nonpolar residues of the active site of STAT3 and S1PR1 are shown in purple and green, respectively. The hydrogen bonds in the sidechain and backbone are shown in red and blue arrows, respectively; the arrows point to the H-acceptor. The π-H interactions are marked with a red line. In the 3D images, the carbon atoms of the compounds are shown in yellow, and other atoms (e.g., oxygen, nitrogen, and sulfur atoms, etc.) are shown in blue. The amino acid residues of the STAT3 and S1PR1 proteins that interacted with compounds are shown in green. The formation of hydrogen bond interactions is shown with a red dotted line
Binding of BTWT constituents to Nrf2
Docking of ML385 to Nrf2 reached the lowest calculated binding energy conformation value of −7.11 kcal/mol, showing that a π-H bond interaction occurred between the benzodioxole group of ML385 and His20 of Nrf2 (Additional file 1: Fig. S1B). The docking simulation of selected components in BTWT to the active site of Nrf2 gave the lowest calculated binding energy conformation values, ranging from -9.14 to -4.66 kcal/mol. Unlike the positive-control ligand ML385, compounds from BTWT did not interact with residue His20 of Nrf2. However, interactions between compounds and other residues occurred; the protonated amine of the isoquinoline ring of palmatine and coptisine made a metal contact with Glu25. No interactions were formed between 23-HA, epiberberine, aesculin, and aesculetin and Nrf2.
Binding of BTWT constituents to PD-1
Nivolumab formed three hydrogen bonds with PD-1, with Asp29 and Gln133 acting as H-acceptor for the hydroxyl group and Ser60 acting as H-acceptor for the ammonia atom of the amide group (Additional file 1: Fig.S1B). The lowest calculated binding energy conformation value was −6.49 kcal/mol. This observed binding mode of nivolumab with Asp29 has been reported in two previous studies [34, 35]. The docking simulation of selected components in BTWT to the active site of PD-1 gave the lowest calculated binding energy conformation values ranging from −6.83–−3.19 kcal/mol. As shown in Fig. 7, only one component of R. pulsatilla, AB4, interacted with PD-1 in a way that was similar to the PD-1–nivolumab interaction. This interaction involved the formation of a hydrogen bond between the hydroxyl group of AB4 and Asp29 acting as a H-acceptor. In addition, the oxygen atom of the methoxy group of jatrorrhizine, epiberberine, and phellodendrine formed hydrogen bonds with Ser60 of PD-1 acting as the H-acceptor.

Molecular docking results of AB4, jatrorrhizine, epiberberine, and phellodendrine in the binding site of PD-1 with the lowest calculated binding energy conformations. In the 2D images, the polar and nonpolar residues of the active site in the PD-1 protein are shown in purple and green, respectively. Hydrogen bonds in the sidechain are shown by red arrows; the arrows point to the H-acceptor. The π-H interactions are marked with a red line and an arene-H label. The π-π interactions are marked with a red line and arene-arene label. The metal contact interactions are shown with a purple line. In the 3D images, the carbon atoms of the compounds are shown in yellow, and other atoms (e.g., oxygen, nitrogen, and sulfur atoms, etc.) are shown in blue. The residues of the PD-1 protein that interacted with the compounds are shown in green. The formation of hydrogen bonds and π-H and π-π interactions are shown by the red dotted line
Binding of BTWT constituents to PD-L1
BMS-202 reached the lowest calculated binding energy conformation value of -8.92 kcal/mol after docking optimization. Four π-H bonds were formed between the phenyl ring of BMS-202 and residues GlnC66, AlaA121, AspC121, and AspA122 of PD-L1. Another π-H bond was formed between the carbon atom of the phenyl ring of BMS-202 and TyrA56 of PD-L1 (Additional file 1: Fig. S1B). Both of these interactions between BMS-202 and PD-L1 have been described in previous studies [44, 45]. In addition, the oxygen atom of the amide group of BMS-202 formed a hydrogen bond with LsyA124 acting as an H-acceptor, and the protonated amine formed a metal contact with AspA122 (Additional file 1: Fig. S2B). About half of the components of BTWT produced the lowest calculated binding energy conformation value that was similar or lower to that of BMS-202; for example, the values of AB4 and berberine were −11.28 and −6.32 kcal/mol, respectively. As shown in Fig. 8, the oxygen atoms of the two hydroxyl groups of AB4 formed two π-H bonds with TyrC56 and TyrA56 (Fig. 8A). In addition, the hydroxyl groups of aesculin formed two hydrogen bonds with TyrC56 and LysA124 acting as H-acceptors (Fig. 8A). Similarly, the phenyl ring of benzopyran-2-one of aesculin formed π-H bonds with residue AspA122 of PD-L1 (Fig. 8A). Furthermore, all six components in R. coptidis and/or C. phellodendri components, which are isoquinoline alkaloids, bound to PD-L1 through interactions with their isoquinoline rings (Fig. 8B); this binding mode was similar to that of BMS-202. Four of these compounds, namely berberine, jatrorrhizine, coptisine, and phellodendrine, formed π-H bonds with residues TyrC56, AlaA121, and/or AspA122 of PD-L1; this interaction occurred through the phenyl ring or the protonated amine of isoquinoline ring of the compounds (Fig. 8B). In addition, palmatine, coptisine, and epiberberine formed metal contacts with AspA122. These results suggest that R. coptidis and/or C. phellodendri contain several potential PD-L1 ligands.

Molecular docking results of AB4, aesculin A, berberine, palmatine, jatrorrhizine, coptisine epiberberine, phellodendrine B with the binding site of PD-L1 with the lowest calculated binding energy conformation. In the 2D images, the polar and nonpolar residues of the active site of the PD-L1 protein are shown in purple and green, respectively. Hydrogen bonds in the sidechain are shown by the red arrows; the arrow points to the H-acceptor. The π-H interactions are marked with a red line and an arene-H label. The π-π interactions are marked with a red line and arene-arene label. The metal contact interactions are shown with a purple line. In the 3D images, the carbon atoms of the compounds are shown in yellow, and other atoms (e.g., oxygen, nitrogen, and sulfur atoms, etc.) are shown in blue. The residues of the PD-L1 protein that interacted with the compounds are shown in green. The formation of hydrogen bonds and π-H and π-π interactions are shown with a red dotted line
Verification of the in-vitro anti-inflammatory activity of the identified immunomodulators from BTWT immunomodulatory components of BTWT
The effects of the BTWT components (showing no cytotoxicity on cultured splenocytes of mice at a concentration of 1 μM, Additional file 1: Fig. S2) on inflammatory responses were investigated using cultured RAW 264.7 cells. LPS stimulation of RAW 264.7 cells increased mRNA and protein levels of TNF-α and IL-6 (Fig. 9A–D). Importantly, the TNF-α and IL-6 levels in RAW 264.7 cells pretreated with the 11 compounds were much lower than those in vehicle-treated controls (Fig. 9A–D). All the compounds significantly reversed the LPS-mediated increase in p-STAT3 expression, and this effect was similar to that produced by tofacitinib (Fig. 9E, F).

The effects of the identified immunomodulators from BTWT on inflammatory responses in vitro. The effects of BTWT components on the production of cytokines in LPS-stimulated RAW264.7 cells were assessed by RT-qPCR (A and B) and ELISA (C and D), and the effects on STAT3 activation were assessed by western blot (E and F). #P < 0.05 and ###P < 0.001 compared with the vehicle control; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the LPS group (n = 3)
