
Abstract
Background
Osteoarthritis is a chronic degenerative joint disease, and increasing evidences suggest that the pathogenic mechanism involves immune system and inflammation.
Aims
The aim of current study was to uncover hub genes linked to immune infiltration in osteoarthritis synovial tissue using comprehensive bioinformatics analysis and experimental confirmation.
Methods
Multiple microarray datasets (GSE55457, GSE55235, GSE12021 and GSE1919) for osteoarthritis in Gene Expression Omnibus database were downloaded for analysis. Differentially expressed genes (DEGs) were identified using Limma package in R software, and immune infiltration was evaluated by CIBERSORT algorithm. Then weighted gene co-expression network analysis (WGCNA) was performed to uncover immune infiltration-associated gene modules. Protein–protein interaction (PPI) network was constructed to select the hub genes, and the tissue distribution of these genes was analyzed using BioGPS database. Finally, the expression pattern of these genes was confirmed by RT-qPCR using clinical samples.
Results
Totally 181 DEGs between osteoarthritis and normal control were screened. Macrophages, mast cells, memory CD4 T cells and B cells accounted for the majority of immune cell composition in synovial tissue. Osteoarthritis synovial showed high abundance of infiltrating resting mast cells, B cells memory and plasma cells. WGCNA screened 93 DEGs related to osteoarthritis immune infiltration. These genes were involved in TNF signaling pathway, IL-17 signaling pathway, response to steroid hormone, glucocorticoid and corticosteroid. Ten hub genes including MYC, JUN, DUSP1, NFKBIA, VEGFA, ATF3, IL-6, PTGS2, IL1B and SOCS3 were selected by using PPI network. Among them, four genes (MYC, JUN, DUSP1 and NFKBIA) specifically expressed in immune system were identified and clinical samples revealed consistent change of these four genes in synovial tissue retrieved from patients with osteoarthritis.
Conclusion
A 4-gene-based diagnostic model was developed, which had well predictive performance in osteoarthritis. MYC, JUN, DUSP1 and NFKBIA might be biomarkers and potential therapeutic targets in osteoarthritis.
Similar content being viewed by others


Introduction
Osteoarthritis is a chronic disease featured by the breakdown of articular cartilage and underlying bone, accompanied by progressive destruction of synovium, ligaments, supporting muscles and meniscus, acting as one of the leading causes of joint disability [1, 2]. Osteoarthritis affects an estimated 240 million people in the world [3], and the risk factors for this disease include age, gender, obesity, joint injury [4], abnormal mechanical load, inadequate nutritional supply and genetic factors [5, 6]. Although osteoarthritis can violate any joints, joints in the hands, knees, hips and spine are the most commonly affected [1,2,3]. The main symptom of osteoarthritis is joint pain, which is not evident at the early stage. Currently, drugs (e.g., paracetamol and naproxen) and physical therapeutic strategies (e.g., exercise and use of a cane) could only alleviate the pain and delay the progression of osteoarthritis to a certain extent [7]. For patients whose quality of life is significantly compromised, joint replacement is recommended. Searching for effective biomarkers is particularly important for the early diagnosis and treatment of osteoarthritis.
Increasing evidences support that osteoarthritis is not merely a cartilage wear-triggered mechanical disorder. The pathogenic mechanism involves the immune system and inflammation [5, 6, 8]. Imaging, pathological and clinical findings indicate that synovial changes occur earlier than the pathological degeneration of cartilage [9,10,11] and synovitis featured by the recruitment of immune cells exists in the whole developmental process of osteoarthritis [12, 13]. Infiltration of activated macrophages and subpopulations of T cells in synovial tissue has been closely linked to the initiation of osteoarthritis [14]. Moreover, the significantly increased T helper type 1 cell and inflammatory cytokines including interferon γ, and interleukin (IL)-2/-10 in synovial fluid contributed to the disease aggravation [15]. Therefore, it is unsurprising that immunological parameters of synovial tissue and dysregulation of gene expression act as crucial discriminants of osteoarthritis.
Immune infiltration as well as associated hub genes and regulatory mechanism has been a hot area of research in cancer [16,17,18]. However, hub genes related to immune infiltration in synovial tissue of osteoarthritis have not been fully identified. There were several studies [16,17,18,19,20] investigating this topic by bioinformatics analysis. For instance, Cai et al. revealed that 14 hub genes (e.g., CCL20, CD44, CX3CR1, CXCL2) related to chemokine and cytokine activity participated in the immune infiltration of osteoarthritis [16]. Hu et al. also conducted bioinformatics analysis and then demonstrated that after stimulation of IL-1β, the expression levels of TCA1, TLR7, MMP9, CXCL10, CXCL13, HLA-DRA and ADIPOQSPP1 were significantly higher in the IL-1β-induced group than in the control group by using in vitro experiments [19]. However, the molecular regulatory network for the hub genes was not constructed in these studies. Therefore, an updated investigation is imperative.
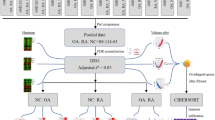
In this study, based on the integrated analysis of several datasets retrieved from the Gene Expression Omnibus (GEO) database, we selected the differentially expressed genes (DEGs) associated with synovial immune infiltration in osteoarthritis using weighted gene co-expression network analysis (WGCNA). Furthermore, several feature genes were uncovered by protein–protein interaction (PPI) analysis and genes tissue distribution and then confirmed by clinical samples.
Methods
Data acquisition and preprocessing
Totally four microarray datasets of osteoarthritis in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) were acquired by using following keyword: osteoarthritis. Data in the GSE55457 dataset (synovial membrane samples from 10 osteoarthritis and 10 control subjects), GSE55235 dataset (synovial membrane samples from 10 osteoarthritis and 10 control subjects) and GSE12021 dataset (synovial membrane samples from 10 osteoarthritis and 10 control subjects) were generated on GPL96 platform. GSE1919 dataset (synovial membrane samples from 5 osteoarthritis and 10 control subjects) was generated on the GPL91 platform. Genes were annotated according to the annotation files provided by corresponding platforms. Probes matched no gene symbols were removed. For multiple probes matched to the same gene symbol, the mean value was used finally. The platform for GSE55457, GSE55235 and GSE12021 datasets was the same, and these three datasets were merged into one dataset for analysis after eliminating the batch effect using the ComBat function provided by the SVA package (version 3.340). GSE1919 dataset was applied as an external validation dataset.
Differential expression analysis
Differentially expression analysis between osteoarthritis vs. normal control was conducted using a t test provided in the Limma package (version 3.10.3), followed by multiple test correction using Benjamini and Hochberg method. DEGs were selected with a cutoff of adjusted p value (adj. p val) < 0.05 and |log(Fold Change(FC))|> 0.585. The ggplot2 package and the pheatmap package were used to visualize the DEGs by drawing the volcano plot and heatmap.
Evaluation of immune cells infiltration
Normalized gene expression data were used to infer the proportions of infiltrating immune cells. The relative abundance of 22 immune cells in each sample was evaluated by the CIBERSORT algorithm referring to the gene expression signature template provided on CIBERSORT official website (https://cibersortx.stanford.edu/, access on July 15, 2022). The parameter was set as perm = 100 and QN = F. The proportion of immune cells in each group was described in the heatmap. Pearson correlation analysis was performed to assess the associations among immune cells.
WGCNA
The calculation principle of WGCNA proposes that genes highly coexpressed in one module are a set of genes associated with the specific disease phenotype. We selected the genes with the absolute deviation of median expression ranked in the top 2000 of all the genes for WGCNA analysis. The adjacency matrix weight parameter power value was first determined to balance the relationship between mean connectivity and scale independence, and the intergenic diverging coefficients were then determined. Next, cluster dendrogram was built to assign genes into diverse modules with the dynamic mixed cutting method, and the minimum number of genes in the module was set as 30. The p value for each gene between groups was calculated using a t test, and the log p value for each gene was defined as gene significance (GS). Module significance (MS) was calculated by the mean value of GS. Finally, the module–trait relationships were computed to reveal the gene modules significantly associated with disease phenotype.
Functional enrichment
Venn analysis was performed to manifest the common genes between DEGs and module genes, and these common genes were considered as key module genes. In order to explore their involved Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and gene ontology annotations, enrichment analysis was conducted utilizing clusterProfiler (version 3.16.0), with cutoff values of gene count ≥ 2 and p < 0.05. Results of functional enrichment analysis were visualized by the ggplot2 package.
PPI network
The interactions among proteins encoded by key module genes were predicted on the basis of the Search Tool for the Retrieval of Interacting Genes (STRING) database (version 11.5, https://cn.string-db.org/, accessed on July 15, 2022), with PPI score set as 0.4 and species as human. The network was then constructed using Cytoscape software (version 3.6.1, Seattle, WA, USA) based on the predicted interactions. The topological property of nodes in the network was analyzed using the cytoHubba algorithm, including degree, maximal clique centrality (MCC), maximum neighborhood component (MNC), betweenness and closeness. The hub genes were defined by the intersection of the top 15 genes for each topological property.
Tissue distribution of hub genes
To identify genes that specifically expressed in the immune system, the distribution of hub genes in different cells or tissues was explored on the basis of the BioGPS database (http://biogps.org/#goto=welcome, accessed on July 15, 2022). The genes specifically expressed in the immune system were considered as feature genes.
Competing endogenous RNAs (ceRNA) network
The circRNA/lncRNA–miRNA–mRNA ceRNA network was established based on the ceRNA theory to screen circRNA, lncRNA and miRNA that can regulate the feature genes. The upstream miRNAs for feature genes were predicted using the miRWalk 3.0 tool (http://mirwalk.umm.uni-heidelberg.de/, access on July 15, 2022), and the miRNA–target pair existed in the miRDB database (http://www.mirdb.org/, accessed on July 15, 2022) with score = 1 was screened. The lncRNAs or circRNAs for miRNAs were predicted based on DIANA-LncBase v.2 (http://www.microrna.gr/LncBase/, accessed on July 15, 2022, score > 0.95, tissue: bone marrow) and ENCORI (https://starbase.sysu.edu.cn/, accessed on July 15, 2022) databases, respectively. The ceRNA network was established by integrating the predicted lncRNAs–miRNAs pairs, circRNAs–miRNAs pairs and miRNA–targets pairs by using Cytoscape.
Validation of feature genes using GSE1919 dataset
The expression data of feature genes in the GSE1919 dataset were used as eigenvalues to construct the support vector machine (SVM) classifier using the e1071 SVM package with the parameters of sigmoid kernel and tenfold cross-validation. In addition, the expression of feature genes in the GSE1919 dataset was visualized using the ggplot2 and ggpubr packages.
RT-qPCR
Synovial tissue samples were collected from three osteoarthritis patients receiving joint replacement and three normal controls receiving arthroscopic exploration in Shandong Provincial Hospital. All participants were informed consent, and this study was approved by the Ethics Committee of Shandong Provincial Hospital. The expression of feature genes was validated using RT-qPCR. Briefly, total RNA isolation from synovial tissues was conducted using TRIZOL reagent, and RNA concentration was determined on a microplate reader. The cDNA was synthesized firstly by reverse transcription with PrimeScript™RT Master Mix (TAKARA, Shiga, Japan). Next, real-time PCR was conducted with Power SYBR Green PCR Master Mix (Thermo, Waltham, MA, USA) on a PCR Amplifier.
Results
DEGs in synovial membrane tissue of osteoarthritis
After eliminating the batch effect, GSE55457, GSE55235 and GSE12021 datasets were merged into one dataset. After normalization, the gene expression of samples in different datasets was at the same level (Figure S1A), and no significant separation among samples in three datasets (Figure S1B), indicating that the merged dataset could be used for subsequent analysis. Differential analysis discovered 181 DEGs between osteoarthritis vs. normal control, of which expression of 67 genes increased, while expression of 114 genes decreased in synovial membrane tissue of osteoarthritis patients than that of normal control (Fig. 1A). The expression pattern of these genes could distinguish samples from osteoarthritis to normal control (Fig. 1B).

Differential expression analysis. A Volcano plot showing the differentially expressed genes between osteoarthritis vs. normal control; B Heatmap showing the expression pattern of genes in between osteoarthritis versus normal control
Immune cells infiltration in osteoarthritis synovial membrane tissue
The relative infiltration abundance of 22 immune cells in synovial membrane tissue was evaluated for each sample. M2 macrophages, resting mast cells, memory CD4 T cells and B cells accounted for the majority of immune cell composition in synovial membrane tissue (Fig. 2A). Pearson correlation analysis revealed that eosinophils were significant correlated with activated dendritic cells (r = 0.75) and neutrophils (r = 0.64). Activated memory CD4 T cells showed strong positive correlations with naive CD4 T cells (r = 0.56) and gamma delta T cells (r = 0.48), while had strong negative correlations with M2 Macrophages (r = − 0.52), resting dendritic cells (r = − 0.45) and activated NK cells (r = − 0.37) (Fig. 2B). Synovial membrane tissue in normal control had a high abundance of resting memory CD4 T cells (p < 0.001), activated NK cells (p = 0.007), activated mast cells (p < 0.001) and eosinophils (p = 0.021), while in osteoarthritis patients, the high infiltrating abundance of resting mast cells (p < 0.001), B cells memory (p = 0.003) and plasma cells (p = 0.001) were observed (Fig. 2C).

Evaluation of immune cells infiltration. A Landscape of immune cells infiltration in synovial membrane tissue of normal control (left) or osteoarthritis patients (right); B Correlation analysis among 22 immune cells in tumor samples; and C Violin plot showing the differences on infiltration abundance of 22 immune cells in synovial membrane tissue of normal control (blue) and osteoarthritis patients (yellow)
Gene modules related to osteoarthritis and immune status
The soft threshold power of 8 (no scale R2 = 0.9) in WGCNA was determined to construct an approximate scale-free network (Fig. 3A). Cluster dendrogram with dynamic mixed cutting method revealed 12 highly connected gene modules (Fig. 3B). Among which, yellow, pink, green and brown modules indicated strong correlations with osteoarthritis (Fig. 3C). Further module–trait relationships analysis showed that yellow and pink modules strongly correlated with both osteoarthritis and multiple immune infiltrating cells (Fig. 3D). Therefore, the genes in yellow and pink modules were considered as osteoarthritis and immune status associated genes.

Weighed gene co-expression network analysis. A The scale independence (left) and mean connectivity (right) for various soft threshold powers; B Cluster dendrogram. Each color branch represents a color-coded module containing a highly interconnected set of genes; C Disease status correlated co-expression modules. X-axis represents different gene modules, and Y-axis represents the overall correlation coefficient between genes in each module and disease status; and D Module–trait relationships and P values for selected traits (osteoarthritis and immune cells)
Identification of feature genes
Venn analysis revealed a total of 93 common genes among two gene modules (yellow and pink) and DEGs (Fig. 4A), and these 93 genes were considered to involve in the occurrence and progression of osteoarthritis. Functional enrichment uncovered that these genes were implicated in TNF signaling pathway, IL-17 signaling pathway and rheumatoid arthritis pathways (Fig. 4B). Multiple biological processes were also enriched, including response to steroid hormone, response to lipopolysaccharide, response to glucocorticoid and response to corticosteroid (Fig. 4C). Based on the STRING database, the interactions among proteins encoded by these genes were analyzed, and 383 interactions involving 67 proteins were obtained (Fig. 5A). Ten hub genes in the intersection of five topological properties were determined (Fig. 5B, Table 1), including MYC, JUN, DUSP1, NFKBIA, VEGFA, ATF3, IL-6, PTGS2, IL1B and SOCS3. Among the ten hub genes, MYC, JUN, DUSP1 and NFKBIA were specifically expressed in immune system based on BioGPS database. Therefore, these four genes were considered as feature genes.

Functional enrichment analysis. A Venn diagram showing the common genes between gene modules and DEGs; B The significantly enriched KEGG pathways for common genes identified from Venn analysis; and C The significantly enriched biological processes terms for common genes identified from Venn analysis

Protein–protein interaction network. A The protein–protein interaction network for common genes identified from Venn analysis. Pink nodes and yellow nodes represent the genes identified from pink or yellow modules in WGCNA; B Identification of hub genes from the network by cytoHubba algorithm
Regulatory network for feature genes
The possible molecular regulation mechanism for feature genes was further explored. As shown in Fig. 6, the ceRNA network consisted of 122 interaction pairs involving 109 nodes, including 12 miRNAs, 43 lncRNAs and 50 circRNAs. For example, lncRNA PCBP1-AS1 might competitively bind to NFKBIA by sponging miR-6802-3p. The top 10 nodes are listed in Table 2, including seven miRNAs (e.g., miR-545-3p, miR-1294, miR-5000-3p), one lncRNA (LOC100190986) and two mRNAs (MYC and JUN). There were more interactions from circRNAs for miR-545-3p, miR-5000-3p and miR-1294, and more interactions from lncRNAs for miR-1827. MYC was targeted by five miRNAs (miR-510-3p, miR-5000-3p, miR-1827, miR-548au-3p and miR-1294). Four miRNAs (miR-6734-3p, miR-3156-3p, miR-6507-3p and miR-4749-3p) targeted JUN.

Regulatory network for feature genes. The lncRNA/circRNA–miRNA–target regulatory network. Yellow nodes represent feature genes; red nodes represent miRNAs; blue nodes represent lncRNAs; and green nodes represent circRNAs
Validation of feature genes using GSE1919 dataset
The expression pattern of four feature genes in osteoarthritis and normal samples is presented in Fig. 7A. Compared to normal control, expression of MYC, JUN, DUSP1 and NFKBIA was down-regulated in osteoarthritis synovial membrane tissue (p value < 0.05).

Validation of feature genes expression in GSE1919 dataset and clinical samples. A Boxplots showing the expression of feature genes in synovial membrane tissue of normal control and osteoarthritis patients. *P < 0.05; **P < 0.01; ***P < 0.01; B Expression of MYC, JUN and DUSP1 in synovial tissues of osteoarthritis patients and normal controls was determined by RT-qPCR. GAPDH was used as internal reference
Expression of feature genes validated by RT-qPCR
For validating the differential expression of feature genes in synovial tissues of osteoarthritis and control subjects, RT-qPCR was performed (Fig. 7B). Consistently, we found that expression of MYC, JUN and DUSP1 was markedly reduced in synovial tissues of osteoarthritis patients than that of normal controls (p value < 0.05). Although there was no statistical significance, the expression of NFKBIA in synovial tissues of osteoarthritis patients showed a reduced trend in comparison with that of normal controls.
Discussion
Diverse immune cells exert crucial roles in the initiation and progression of osteoarthritis. Prompt application of diagnostic and therapeutic biomarkers would dramatically improve the prognosis of osteoarthritic patients, but currently few molecular candidates fit the criteria of high sensitivity and specificity. In this study, we characterized the differences in terms of gene expression and immune cell infiltration in osteoarthritis synovial tissue compared to normal control. Further WGCNA screened 93 differentially expressed genes associated with osteoarthritis and immune status, and these genes were involved in immuno-inflammatory related pathways (e.g., TNF signaling pathway and IL-17 signaling pathway) and biological response processes (e.g., response to steroid hormone/glucocorticoid/corticosteroid). Finally, four feature genes, including MYC, JUN, DUSP1 and NFKBIA were identified, which had well predictive performance in osteoarthritis.
Immune infiltration evaluation indicated that macrophages, mast cells, memory CD4 T cells and B cells accounted for the majority of immune cell composition in synovial tissue. Osteoarthritis patients showed a relative high abundance of infiltrating resting mast cells, memory B cells and plasma cells. Increasing evidences have demonstrated that osteoarthritis is not non-inflammatory arthritis as previously described, but a disease that involves continuous low-level inflammation and activation of innate inflammatory pathways [21]. Synovial macrophages are the primary inflammatory and immune cells, and a large number of activated macrophages are found in synovial tissues of patients with osteoarthritis at different stages [22]. M1 macrophages account for the primary subgroup involving the inflammatory factors release, cartilage degradation and osteophyte formation in osteoarthritis, while M2 macrophages produce anti-inflammatory cytokines, showing a protective effect in osteoarthritis [22, 23]. The number of mast cells in synovium was markedly increased in osteoarthritic patients than control, and closely correlated with synovial inflammation and disease severity [24]. Elevated mast cell infiltration could amplify the inflammatory pathology and aggravate structural damage of joints in osteoarthritis [25, 26]. In addition, B cells and plasma cells were also detected in synovial tissue or synovial fluid of osteoarthritic patients, of which B cells were a source of IL-10, while plasma cells could secrete IL-6 [27, 28]. These findings suggested that targeting synovial immune cells might be a potential therapeutic strategy for osteoarthritic patients.
Based on WGCNA, we further revealed a set of genes associated with osteoarthritis and immune status, which were involved in the TNF signaling pathway, and IL-17 signaling pathway. Synovial cells can be divided into two types [29]. Type A includes macrophage-like cells with phagocytic function, while type B is fibroblast-like synoviocytes (FLS), which mainly provide lubrication and nutrition for joints by secreting hyaluronic acid [29, 30]. TNF-α could induce the production of various cytokines (matrix metalloproteinases (MMPs) and IL-34) to participate in the inflammatory response of osteoarthritis by activating different signals [31,32,33,34]. IL-17 is a pleiotropic inflammatory cytokine involved in osteoarthritis, and blocking the IL-17 signaling pathway can effectively relieve pain for patients [35, 36]. IL-17-mediated inflammation leads to mitochondrial dysfunction of FLS, which in turn stimulates the infiltration of inflammatory lymphocytes and autophagic apoptosis of FLS [37]. In the osteoarthritis mice model, IL-17 facilitates cartilage destruction and nociceptive properties by regulating inflammatory mediators [38]. In addition, these DEGs were also enriched in response to steroid hormone, glucocorticoid and corticosteroid. Glucocorticoids have been used for the treatment of osteoarthritis [39, 40]. These findings suggested that the screened genes played important roles in osteoarthritis.
Next, by further analysis of ten hub genes (MYC, JUN, DUSP1, NFKBIA, VEGFA, ATF3, IL-6, PTGS2, IL1B and SOCS3), we discovered that MYC, JUN, DUSP1 and NFKBIA were specifically expressed in the immune system and selected as biomarkers and potential therapeutic targets in osteoarthritis. Inhibition of the MYC gene promoted cell proliferation and repressed cell apoptosis, and expression of MMP-13, IL-6 and TNF-α in IL-1β-induced rat chondrocytes [41]. JUN has been reported to stimulate the apoptosis of chondrocytes by increasing the level of PUMA (a pro-apoptotic factor) [42] and BIM [43], serving an important role in osteoarthritis. Jian et al. demonstrated that expression of DUSP1 was reduced in synovial tissues facet joint osteoarthritis patients and IL-1β-induced FLS, and overexpression of DUSP1 inhibited the proliferation and inflammatory response in FLS [44]. DUSP1 has a protective effect on osteoarthritis by intercepting the expression of MMP-13 and activating the MAPK pathway [45]. NFKBIA is a member of the NFKB inhibitor family, which prevents inflammatory responses-related NFKB/REL complexes by interacting with REL [46]. NFKBIA was involved in the inflammatory effect of FLS, contributing to an elevated risk of hip osteoarthritis [46, 47].
LncRNA, circRNA and miRNA are non-coding RNAs (ncRNAs) that constitute approximately 90% of the RNA transcripts of the human genome. Despite lacking protein-coding potential, ncRNAs participated in a variety of pathological processes of osteoarthritis by regulating gene transcription and protein translation, serving as biological targets in the prevention, diagnosis and treatment of osteoarthritis [48, 49]. Therefore, the lncRNA/circRNA–miRNA–mRNA regulatory network for these four hub genes was constructed and several hub ncRNAs such as miR-545-3p, miR-1294 and miR-510-3p were selected. Liu et al. have demonstrated the effect of miR-510-3p in inhibiting the progression of osteoarthritis [50]. MiR-545-3p and miR-1294 have not been investigated in osteoarthritis, but studies have confirmed their effectiveness in regulating inflammation. For example, the dysregulation of miR-545 is associated with elevated expression of inflammatory cytokines and abnormal activation of macrophage in cirrhosis [51]. Besides, miR-545 could affect the inflammatory response in cardiomyocytes by targeting CXCL16 [52]. Upregulation of miR-1294 inhibits inflammation in atopic dermatitis by suppressing NF-κB pathway activation [53] and promotes the development of autoimmune disorders by regulating the release of pro-inflammatory cytokines [54].
Despite these findings, there remain some limitations in this study. First, only qPCR was performed to validate the gene expression at the transcriptomic level for three samples in each group. In addition, the biological function and the clinical correlations of these feature genes in the development of osteoarthritis have not been investigated. Second, the diagnostic value of these feature genes was only verified based on a small dataset, which undermines the power of the validation. Last but not least, the hub lncRNAs/circRNAs/miRNAs were predicted by using multiple bioinformatics analysis methods, but the role of these identified ncRNAs in the progression of osteoarthritis remains unclear. It should be admitted that the experimental validation section of this investigation is very elementary. However, findings of the bioinformatics analysis provide multiple directions for research about the pathological mechanism of osteoarthritis, which will be tapped in the future.
Conclusion
In conclusion, gene expression and immune infiltration in osteoarthritis synovial tissue were characterized in this study. A set of genes related to osteoarthritis immune infiltration was identified, which might play essential roles in the development and progression of osteoarthritis. Four genes, MYC, JUN, DUSP1 and NFKBIA, were selected as potential diagnostic biomarkers in osteoarthritis. This study provided novel clues for the early diagnosis and treatment of osteoarthritis.
Availability of data and materials
All data analyzed during this study are included in this published article.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- WGCNA:
-
Weighted gene co-expression network analysis
- PPI:
-
Protein–protein interaction
- DEGs:
-
Differentially expressed genes
- FC:
-
Fold change
- GS:
-
Gene significance
- MS:
-
Module significance
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MCC:
-
Maximal clique centrality
- MNC:
-
Maximum neighborhood component
- SVM:
-
Support vector machine
References
Abramoff B, Caldera FE. Osteoarthritis: pathology, diagnosis, and treatment options. Med Clin N Am. 2020;104:293–311. https://doi.org/10.1016/j.mcna.2019.10.007.
Barnett R. Osteoarthritis. Lancet (London, England). 2018;391:1985. https://doi.org/10.1016/s0140-6736(18)31064-x.
Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA. 2021;325:568–78. https://doi.org/10.1001/jama.2020.22171.
Parekh SM, Fernandes GS, Moses JP, et al. Risk factors for knee osteoarthritis in retired professional footballers: a cross-sectional study. Clin J Sport Med Off J Can Acad Sport Med. 2021;31:281–8. https://doi.org/10.1097/jsm.0000000000000742.
Allen KD, Thoma LM, Golightly YM. Epidemiology of osteoarthritis. Osteoarthr Cartil. 2022;30:184–95. https://doi.org/10.1016/j.joca.2021.04.020.
Ratneswaran A, Kapoor M. Osteoarthritis year in review: genetics, genomics, epigenetics. Osteoarthr Cartil. 2021;29:151–60. https://doi.org/10.1016/j.joca.2020.11.003.
Quicke JG, Conaghan PG, Corp N, et al. Osteoarthritis year in review 2021: epidemiology and therapy. Osteoarthr Cartil. 2022;30:196–206. https://doi.org/10.1016/j.joca.2021.10.003.
Woodell-May JE, Sommerfeld SD. Role of inflammation and the immune system in the progression of osteoarthritis. J Orthop Res Off Publ Orthop Res Soc. 2020;38:253–7. https://doi.org/10.1002/jor.24457.
Scanzello CR, McKeon B, Swaim BH, et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum. 2011;63:391–400. https://doi.org/10.1002/art.30137.
Guermazi A, Roemer FW, Hayashi D, et al. Assessment of synovitis with contrast-enhanced MRI using a whole-joint semiquantitative scoring system in people with, or at high risk of, knee osteoarthritis: the MOST study. Ann Rheum Dis. 2011;70:805–11. https://doi.org/10.1136/ard.2010.139618.
Roemer FW, Guermazi A, Felson DT, et al. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: the MOST study. Ann Rheum Dis. 2011;70:1804–9. https://doi.org/10.1136/ard.2011.150243.
Miller RJ, Malfait AM, Miller RE. The innate immune response as a mediator of osteoarthritis pain. Osteoarthr Cartil. 2020;28:562–71. https://doi.org/10.1016/j.joca.2019.11.006.
Deligne C, Casulli S, Pigenet A, et al. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthr Cartil. 2015;23:1843–52. https://doi.org/10.1016/j.joca.2014.12.007.
Lopes EBP, Filiberti A, Husain SA, et al. Immune contributions to osteoarthritis. Curr Osteoporos Rep. 2017;15:593–600. https://doi.org/10.1007/s11914-017-0411-y.
Rosshirt N, Hagmann S, Tripel E, et al. A predominant Th1 polarization is present in synovial fluid of end-stage osteoarthritic knee joints: analysis of peripheral blood, synovial fluid and synovial membrane. Clin Exp Immunol. 2019;195:395–406. https://doi.org/10.1111/cei.13230.
Huang X, Zhou S, Toth J, et al. Cuproptosis-related gene index: a predictor for pancreatic cancer prognosis, immunotherapy efficacy, and chemosensitivity. Front Immunol. 2022;13:978865. https://doi.org/10.3389/fimmu.2022.978865.
**e J, Zheng S, Zou Y, et al. Turning up a new pattern: identification of cancer-associated fibroblast-related clusters in TNBC. Front Immunol. 2022;13:1022147. https://doi.org/10.3389/fimmu.2022.1022147.
Wang Q, Huang X, Zhou S, et al. IL1RN and PRRX1 as a prognostic biomarker correlated with immune infiltrates in colorectal cancer: evidence from bioinformatic analysis. Int J Genomics. 2022;2022:2723264. https://doi.org/10.1155/2022/2723264.
Cai W, Li H, Zhang Y, et al. Identification of key biomarkers and immune infiltration in the synovial tissue of osteoarthritis by bioinformatics analysis. PeerJ. 2020;8:e8390. https://doi.org/10.7717/peerj.8390.
Hu X, Ni S, Zhao K, et al. Bioinformatics-led discovery of osteoarthritis biomarkers and inflammatory infiltrates. Front Immunol. 2022;13:871008. https://doi.org/10.3389/fimmu.2022.871008.
Griffin TM, Scanzello CR. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin Exp Rheumatol. 2019;37(Suppl 120):57–63.
Thomson A, Hilkens CMU. Synovial macrophages in osteoarthritis: the key to understanding pathogenesis? Front Immunol. 2021;12:678757. https://doi.org/10.3389/fimmu.2021.678757.
Hu Y, Gui Z, Zhou Y, et al. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic Biol Med. 2019;145:146–60. https://doi.org/10.1016/j.freeradbiomed.2019.09.024.
Farinelli L, Aquili A, Mattioli-Belmonte M, et al. Synovial mast cells from knee and hip osteoarthritis: histological study and clinical correlations. J Exp Orthop. 2022;9:13. https://doi.org/10.1186/s40634-022-00446-2.
de Lange-Brokaar BJ, Kloppenburg M, Andersen SN, et al. Characterization of synovial mast cells in knee osteoarthritis: association with clinical parameters. Osteoarthr Cartil. 2016;24:664–71. https://doi.org/10.1016/j.joca.2015.11.011.
Kulkarni P, Harsulkar A, Märtson AG, et al. Mast cells differentiated in synovial fluid and resident in osteophytes exalt the inflammatory pathology of osteoarthritis. Int J Mol Sci. 2022;23:541. https://doi.org/10.3390/ijms23010541.
Sun H, Zhang Y, Song W, et al. IgM(+)CD27(+) B cells possessed regulatory function and represented the main source of B cell-derived IL-10 in the synovial fluid of osteoarthritis patients. Hum Immunol. 2019;80:263–9. https://doi.org/10.1016/j.humimm.2019.02.007.
Doss F, Menard J, Hauschild M, et al. Elevated IL-6 levels in the synovial fluid of osteoarthritis patients stem from plasma cells. Scand J Rheumatol. 2007;36:136–9. https://doi.org/10.1080/03009740701250785.
Falconer J, Murphy AN, Young SP, et al. Review: Synovial cell metabolism and chronic inflammation in rheumatoid arthritis. Arthritis Rheumatol (Hoboken, NJ). 2018;70:984–99. https://doi.org/10.1002/art.40504.
Maglaviceanu A, Wu B, Kapoor M. Fibroblast-like synoviocytes: role in synovial fibrosis associated with osteoarthritis. Wound Repair Regen. 2021;29:642–9. https://doi.org/10.1111/wrr.12939.
Zhang Q, Ouyang Z, Song X, et al. Epigenetic modifications of tumor necrosis factor-alpha in joint cartilage tissue from osteoarthritis patients - CONSORT. Medicine. 2021;100:e27868. https://doi.org/10.1097/md.0000000000027868.
Malemud CJ. Matrix metalloproteinases and synovial joint pathology. Prog Mol Biol Transl Sci. 2017;148:305–25. https://doi.org/10.1016/bs.pmbts.2017.03.003.
Özler K, Aktaş E, Atay Ç, et al. Serum and knee synovial fluid matrix metalloproteinase-13 and tumor necrosis factor-alpha levels in patients with late-stage osteoarthritis. Acta Orthop Traumatol Turcica. 2016;50:356–61. https://doi.org/10.3944/aott.2015.15.0115.
Udomsinprasert W, **awath A, Teerawattanapong N, et al. Interleukin-34 overexpression mediated through tumor necrosis factor-alpha reflects severity of synovitis in knee osteoarthritis. Sci Rep. 2020;10:7987. https://doi.org/10.1038/s41598-020-64932-2.
Liu Y, Peng H, Meng Z, et al. Correlation of IL-17 level in synovia and severity of knee osteoarthritis. Med Sci Monit Int Med J Exp Clin Res. 2015;21:1732–6. https://doi.org/10.12659/msm.893771.
Faust HJ, Zhang H, Han J, et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Investig. 2020;130:5493–507. https://doi.org/10.1172/jci134091.
Kim EK, Kwon JE, Lee SY, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis 2017; 8:e2565.
Na HS, Park JS, Cho KH, et al. Interleukin-1-interleukin-17 signaling axis induces cartilage destruction and promotes experimental osteoarthritis. Front Immunol. 2020;11:730. https://doi.org/10.3389/fimmu.2020.00730.
He WW, Kuang MJ, Zhao J, et al. Efficacy and safety of intraarticular hyaluronic acid and corticosteroid for knee osteoarthritis: a meta-analysis. Int J Surg (London, England). 2017;39:95–103. https://doi.org/10.1016/j.ijsu.2017.01.087.
Samuels J, Pillinger MH, Jevsevar D, et al. Critical appraisal of intra-articular glucocorticoid injections for symptomatic osteoarthritis of the knee. Osteoarthr Cartil. 2021;29:8–16. https://doi.org/10.1016/j.joca.2020.09.001.
Zou J, Li XL, Shi ZM, et al. Effects of C-myc gene silencing on interleukin-1β-induced rat chondrocyte cell proliferation, apoptosis and cytokine expression. J Bone Miner Metab. 2018;36:286–96. https://doi.org/10.1007/s00774-017-0845-4.
Lu H, Hou G, Zhang Y, et al. c-Jun transactivates Puma gene expression to promote osteoarthritis. Mol Med Rep. 2014;9:1606–12. https://doi.org/10.3892/mmr.2014.1981.
Ye Z, Chen Y, Zhang R, et al. c-Jun N-terminal kinase - c-Jun pathway transactivates Bim to promote osteoarthritis. Can J Physiol Pharmacol. 2014;92:132–9. https://doi.org/10.1139/cjpp-2013-0228.
Jian S, Luo D, Wang Y, et al. MiR-337-3p confers protective effect on facet joint osteoarthritis by targeting SKP2 to inhibit DUSP1 ubiquitination and inactivate MAPK pathway. Cell Biol Toxicol. 2021. https://doi.org/10.1007/s10565-021-09665-2.
Peng HZ, Yun Z, Wang W, et al. Dual specificity phosphatase 1 has a protective role in osteoarthritis fibroblast-like synoviocytes via inhibition of the MAPK signaling pathway. Mol Med Rep. 2017;16:8441–7. https://doi.org/10.3892/mmr.2017.7617.
Tang H, Cheng Z, Ma W, et al. TLR10 and NFKBIA contributed to the risk of hip osteoarthritis: systematic evaluation based on Han Chinese population. Sci Rep. 2018;8:10243. https://doi.org/10.1038/s41598-018-28597-2.
Cai P, Jiang T, Li B, et al. Comparison of rheumatoid arthritis (RA) and osteoarthritis (OA) based on microarray profiles of human joint fibroblast-like synoviocytes. Cell Biochem Funct. 2019;37:31–41. https://doi.org/10.1002/cbf.3370.
Wu Y, Lu X, Shen B, et al. The therapeutic potential and role of miRNA, lncRNA, and circRNA in osteoarthritis. Curr Gene Ther. 2019;19:255–63. https://doi.org/10.2174/1566523219666190716092203.
Kong H, Sun ML, Zhang XA, et al. Crosstalk among circRNA/lncRNA, miRNA, and mRNA in osteoarthritis. Front Cell Dev Biol 2021, 9:774370. https://doi.org/10.3389/fcell.2021.774370.
Liu Y, Yang Y, Ding L, et al. LncRNA MIR4435-2HG inhibits the progression of osteoarthritis through miR-510-3p sponging. Exp Ther Med. 2020;20:1693–701. https://doi.org/10.3892/etm.2020.8841.
Liu X, Li C, Zhu J, et al. Dysregulation of FTX/miR-545 signaling pathway downregulates Tim-3 and is responsible for the abnormal activation of macrophage in cirrhosis. J Cell Biochem. 2019;120:2336–46. https://doi.org/10.1002/jcb.27562.
Liang FQ, Gao JY, Liu JW. C-X-C motif chemokine 16, modulated by microRNA-545, aggravates myocardial damage and affects the inflammatory responses in myocardial infarction. Hum Genomics. 2021;15:15. https://doi.org/10.1186/s40246-021-00314-7.
Yan C, Ying J, Lu W, et al. MiR-1294 suppresses ROS-dependent inflammatory response in atopic dermatitis via restraining STAT3/NF-κB pathway. Cell Immunol 2022, 371:104452. https://doi.org/10.1016/j.cellimm.2021.104452.
Pluta L, Yousefi B, Damania B, et al. Endosomal TLR-8 senses microRNA-1294 resulting in the production of NFḱB dependent cytokines. Front Immunol. 2019;10:2860. https://doi.org/10.3389/fimmu.2019.02860.
Acknowledgements
We thank the authors of the included studies for their help.
Funding
This work was partially supported by the Young Taishan Scholars Program of Shandong Province (QZ, Grant No. tsqn201909183), the Academic promotion programme of Shandong First Medical University (QZ, Grant No. 2020RC008) and the Natural Science Foundation of Shandong Province (QZ, Grant No. ZR2020QH072).
Author information
Authors and Affiliations
Contributions
QZ and CS analyzed and interpreted data. QZ, CS and XL collected data and performed WGCNA and PPI analysis. CZ and CM performed experimental investigation. QZ was major contributor in the manuscript writing. JF designed the study and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Principal component analysis and normalization of samples. A The distribution of expression of 27 samples involving principal component analysis (PCA) for confirming biological variability between different samples; B The distribution of expression of 30 samples before and after normalization.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Q., Sun, C., Liu, X. et al. Mechanism of immune infiltration in synovial tissue of osteoarthritis: a gene expression-based study. J Orthop Surg Res 18, 58 (2023). https://doi.org/10.1186/s13018-023-03541-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-023-03541-x