
Abstract
Background
The construction and application of synthetic genetic circuits is frequently improved if gene expression can be orthogonally controlled, relative to the host. In plants, orthogonality can be achieved via the use of CRISPR-based transcription factors that are programmed to act on natural or synthetic promoters. The construction of complex gene circuits can require multiple, orthogonal regulatory interactions, and this in turn requires that the full programmability of CRISPR elements be adapted to non-natural and non-standard promoters that have few constraints on their design. Therefore, we have developed synthetic promoter elements in which regions upstream of the minimal 35S CaMV promoter are designed from scratch to interact via programmed gRNAs with dCas9 fusions that allow activation of gene expression.
Results
A panel of three, mutually orthogonal promoters that can be acted on by artificial gRNAs bound by CRISPR regulators were designed. Guide RNA expression targeting these promoters was in turn controlled by either Pol III (U6) or ethylene-inducible Pol II promoters, implementing for the first time a fully artificial Orthogonal Control System (OCS). Following demonstration of the complete orthogonality of the designs, the OCS was tied to cellular metabolism by putting gRNA expression under the control of an endogenous plant signaling molecule, ethylene. The ability to form complex circuitry was demonstrated via the ethylene-driven, ratiometric expression of fluorescent proteins in single plants.
Conclusions
The design of synthetic promoters is highly generalizable to large tracts of sequence space, allowing Orthogonal Control Systems of increasing complexity to potentially be generated at will. The ability to tie in several different basal features of plant molecular biology (Pol II and Pol III promoters, ethylene regulation) to the OCS demonstrates multiple opportunities for engineering at the system level. Moreover, given the fungibility of the core 35S CaMV promoter elements, the derived synthetic promoters can potentially be utilized across a variety of plant species.
Similar content being viewed by others

Introduction
The field of synthetic biology aims to revolutionize biotechnology by rationally engineering living organisms [1,2,3,4,5,6]. One aspect of rational engineering is to embed biological organisms with complex information processing systems that can be used to control phenotypes [2, 3, 7, 8], often via synthetic gene circuits that can predictability regulate and tune expression of endogenous as well as transgenes [4, 9,10,11].
However the performance of such synthetic genetic circuits is often plagued by unwanted interactions between the circuit components and the host regulatory system, which can lead to loss of circuit function [10]. These unprogrammed interactions can be mitigated via the design and use of genetic parts that have minimal cross-talk with the host, creating orthogonal regulatory or orthogonal control systems (OCS) that can further serve as the basis for constructing complex genetic programs with predictable behaviors. In the last two decades an increasing number of well-characterized genetic parts have been combined in circuits capable of complex dynamic behaviors, including bi-stable switches, oscillators, pulse generators, Boolean-complete logic gates [7, 12,13,14,15]. While OCS and the circuits that comprise them were initially characterized in microbial hosts, more recently a significant fraction of them have been constructed and characterized in eukaryotic hosts such as yeast and mammalian cells [12, 16,17,18,19]. More recently, synthetic transcriptional control elements have begun to be characterized in plants [20,21,22].
While a variety of artificial plant transcription factors containing diverse DNA binding domains and plant-specific regulatory sequences are known [20, 22], orthogonal control requires more programmable DNA binding domains and modular regulatory domains [20, 22,23,24]. To this end, we describe an alternate strategy for the construction of orthogonal transcriptional regulatory elements in plants, powered by a single universal transcriptional factor – dCas9:VP64 which has been shown to work in a wide variety of eukaryotic species, including plants [16, 7, 9, 28, 29]. Because the overlaps for assemblies can be modularly specified, multiple parts can be assembled sequentially in a single tube reaction.
While a Golden-Gate framework was previously described for the construction of plant expression vectors [30], here we used the highly optimized modular cloning (MoClo) framework, instantiated as a yeast toolkit (YTK) as the basis of our architecture [28]. Recently, beyond yeast expression vectors, this framework has been adapted for the construction of a mammalian toolkit (MTK) [9]. Along with both YTK and MTK, a plant toolkit based on the YTK architecture will prove essential for seamlessly porting parts and circuits across diverse eukaryotes. Briefly, in this framework the entire vector is divided into particular ‘part’ types flanked by BsaI restriction sites followed by a unique ligation site. Promoters, genes and terminators are generally categorized into Type 2, 3 and 4 parts respectively where each part type has a unique overhang that dictates the compatibility between part types [9, 28] (Fig. 1A, Additional file 1: Fig. S1A). This preserves the architecture of each transcriptional unit (promoter-gene-terminator). For the assembly of multiple transcriptional units (TU), each transcriptional unit is first cloned into an ‘intermediate’ vector flanked by connector sequences that dictate the order of the TUs to be stitched together. By using appropriate connectors, each TU can be further assembled into a final expression vector in a single pot reaction (Additional file 1: Fig. S1B) [20]. This modular approach enables rapid assembly of increasingly complex genetic circuits comprised of multiple transcriptional units.

Schematic overview of the design-build-test cycle A Genetic elements such as promoters, genes and terminators are encoded as modular parts consisting of BsaI recognition sites flanked by specific overhangs to ensure the hierarchical assembly of transcriptional units. Once assembled, the constructs are transformed into Agrobacterium and the reporter expression is characterized in Nicotiana benthamiana leaf infiltrates B Design of the shuttle vector backbone used for the assembly of constructs and subsequent propagation in Agrobacterium
Since Agrobacterium-based transformation has been the staple for plant genetic engineering for decades [31], we used compatible vectors as the basis for our framework, and designed and constructed three YTK-compatible shuttle vectors. Each expression vector contains the pVS1 replicon (an Agrobacterium origin of replication – OriV and two supporting proteins—RepA and StaA) and pBR22 origin for propagation in Agrobacterium and E.coli respectively, and a common antibiotic selection cassette (KanR) that has been shown to be functional in both species (Fig. 1B, Materials and Methods) [29, 30]. The three constructs otherwise differed in the plant selection marker—BASTA, hygromycin, and kanamycin. The resistance markers were expressed from the Nos promoter and also contained a Nos terminator [30] (Fig. 1B). The backbone also contains a GFP drop-out cassette that allows easy identification of correct assemblies, which should appear as colonies that lack fluorescence [9, 28] (Fig. 1B).
Fluorescence and luminescence reporters are frequently used to study protein localization and interaction in plants and animals [32]. To provide these useful reporter parts in the context of our system, we cloned the strong promoter from Cauliflower mosaic virus (35S) as a Type 2 part and its corresponding terminator as a Type 4 part [33, 34]. These parts can be matched with a number of fluorescent reporter genes (GFP, BFP, YFP and RFP) all as Type 3 parts for robust reporter expression. Combinations of these proteins can also potentially be used for BIFC (Bimolecular Fluorescence Complementation) [35]. Similarly, luciferase is commonly used in plant molecular biology to study circadian rhythm [36], test the spatiotemporal activities of regulatory elements [50, 51].
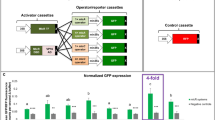
To proof the ribozyme processed gRNA constructs, OCS circuits were assembled where gRNAs were either expressed from a U6 promoter (OCS 1–1) or the 35S promoter (OCS 1–5), and could subsequently activate the transcription and expression of reporter genes (YFP) (Fig. 5A). For both OCS circuits, downstream reporter gene expression was observed, at similar levels (Fig. 5B). The specific levels of gRNA obtained in each case were analyzed using qRT-PCR (Fig. 5C and D), and as expected the level of gRNA from the strong Pol II (35S) driven expression was higher than those obtained with the U6 promoter while similar levels of reporter expression were observed for both cases, thus demonstrating that this Pol II driven gRNA expression strategy can be effectively used for OCS activation (Fig. 5E). For both these constructs the expression of hdCas9 (human codon optimized dCas9) was also confirmed via Western blot analysis (Additional file 1: Fig. S2).

Design and characterization of gRNA expression modules under the control of Pol II promoters. A OCS1-1 circuit generates RNA using U6 (Pol III) promoter while OCS1-5 circuit generates gRNA using 35S (Pol II) promoter flanked by self-cleaving ribozymes – HammerHead (HHR) and Hepatitis Delta Virus (HDV). B Fluorescence microscope images showing Agrobacterium mediated transient expression of OCS constructs with two modalities of gRNA expression (OCS1-1 and OCS1-5). Control images were taken without dCAS9-VP64 expression. Scale bars: 200 μm C and D. Quantification of the gRNA-1 expression in OCS constructs (OCS 1–1 (C) and OCS 1–5 (D)) using qPCR relative to 5S rRNA. Error bars: S.D. (n = 3, independent replicates) E. Relative integrated density of each fluorescence signal (shown in panel B). Integrated density was measured using image J software and normalized to that of the control (con;—dCas9-VP64). Error bars: S.D. (n = 3, independent replicates). Asterisks indicate statistical significance in a student t-test (P < 0.05). NS: not significant
In order to demonstrate that the Pol II-driven gRNAs could be used as part of an inducible OCS, we used the well-characterized synthetic EBS promoter containing the EIN3 binding sites [52], and placed YFP under the downstream control of the ATF (via pATF-1) (Fig. 6A). This circuit (OCS1-9) should be inducible by the volatile organic compound (VOC) ethylene, which is produced from its precursor ACC (1-aminocyclopropane-1-carboxylic acid). Time-dependent expression of YFP is observed in response to 10 μM ACC induction (Fig. 6B). Both the gRNA-1 and YFP expression levels were quantified before and after induction by qRT-PCR, a maximum of threefold induction was observed for both cases (Fig. 6C and D). Thus, this demonstrates that the activity from synthetic promoters can be controlled via the selective expression of the corresponding gRNAs.

Characterization of an ethylene inducible orthogonal control system. A OCS1-9 circuit (gRNA-1 is expressed by ethylene inducible EBS promoter) B Time course fluorescence microscope images showing Agrobacterium mediated transient expression of OCS1-9 in Nicotiana benthamiana leaves after induction with 10 μM ACC. Scale bars: 200 μm C and D qPCR quantification of gRNA-1 (C) and YFP (D) expression before and after induction with ACC, where both show similar levels of induction demonstrating that the relative change in gRNA-1 expression (ethylene induction) results in the differential activation from the pATF-1 promoter. Error bars: S.D. (n = 3, independent replicates), Asterisks indicate statistical significance in a student t-test (P < 0.05)
Construction of a panel of mutually orthogonal synthetic promoters
Lack of multiplexed control of transgenes has been a major factor limiting the development of synthetic circuits in plants [5, 6]. Multiplexed regulation in turn requires a panel of mutually orthogonal promoters and control elements that can operate simultaneously [5, 6]. Our strategy for synthetic promoter design leads to expression cassettes that are mutually orthogonal, while the control of the synthetic promoters via the regulation of gRNA expression for dCas9:VP64 provides orthogonality between the OCS and host regulation. By simply minimizing homology between gRNAs, we constructed two additional promoters similar to the architecture of pATF-1, in which gRNA binding sites were followed by a minimal 35S promoter (pATF-3 and pATF-4). The orthogonality of these promoters was assayed by assembling expression constructs in which each synthetic promoter controlled the production of a unique fluorescent reporter (pATF-1: YFP, pATF-3: RFP and pATF-4: BFP). The respective gRNAs (gRNA-1, gRNA-3 and gRNA-4) were separately transcribed from a U6 promoter (Fig. 7A). When expression constructs were infiltrated into Nicotiana benthamiana, each of the synthetic promoters was specifically upregulated only when its corresponding gRNA was expressed; no background was detected from the remaining two synthetic promoters. (Fig. 7B and C).

Degree of orthogonality of synthetic promoters. A OCS circuit containing all three synthetic promoters (pATF-1, pATF-3 and pATF-4) driving three different reporter genes namely YFP, BFP and RFP respectively with a single gRNA expressed one at a time under the control of U6 promoter. B Fluorescence microscope images showing Agrobacterium mediated transient expression of OCS constructs in Nicotiana benthamiana leaves. Scale bars: 200 μm C As observed from the fluorescence images, only the specific gRNA:pATF pair is active, thus demonstrating that the synthetic promoters are mutually orthogonal Relative integrated density of each fluorescence signal (shown in panel B). Integrated density was measured by image J software and normalized to the highest value. Error bars: S.D. (n = 3, independent replicates). Asterisks indicate statistical significance in a student t-test (P < 0.05)
Construction of complex ratiometric circuits
Now that we have a suite of mutually orthogonal promoters, we sought to construct simple circuits where the activity of each promoter could be independently controlled. Three separate reporter proteins were used to simultaneously monitor the activity of two promoters: pATF-1 with YFP, while both RFP and BFP were under the control of the pATF-3. By leveraging the designed, orthogonal behavior of these promoters it proved possible to construct a ratiometric circuit wherein the activity of pATF-1, and hence YFP expression, was under the control of ethylene (via ACC), while pATF-3 constitutively drove the expression of RFP and BFP (Fig. 8A). As expected, the addition of 10 μM ACC, induced the expression of YFP from the pATF-1 promoter (threefold), while the expression of the other reporters remained constant (Fig. 8B and C). The ratiometric response was further validated by qRT-PCR; pATF-1 was induced threefold following a similar increase in expression of gRNA-1 while there were no changes observed in the transcription of the other two reporter genes (Fig. 8B and C). The predictable behavior of the designed, artificial control elements in the ratiometric circuit is one of the first examples of complex circuitry to be described in plants, and demonstrates uniquely how natural metabolism and regulatory circuitry can be interfaced with free-standing orthogonal control systems.

Design and characterization of a ratiometric circuit. A OSC3-5 contains YFP which is inducible by ACC (pATF-1), while BFP and RFP are constitutively expressed under the control of pATF-3 via the constitutive expression of gRNA-3. B Fluorescence microscope images showing Agrobacterium mediated transient expression of the ratiometric OCS construct (OCS3-5) in Nicotiana benthamiana leaves with or without 10 μM ACC. Scale bars: 200 μm C qPCR quantification of YFP, BFP and RFP shows that YFP is induced after the treatment with ACC while the expression of BFP and RFP remains unchanged before or after ACC induction. Error bars: S.D. (n = 4, independent replicates). An asterisk indicates statistical significance in a student t-test (P < 0.05)
Discussion
Transcriptional orthogonality is one of the bedrocks for circuit construction in synthetic biology, and generally serves as the basis for the bottom-up construction of complex circuitry for predictable dynamics [7, 10, 17]. For eukaryotes the construction of multiple promoter elements is hindered by the typically complex regulatory sequences that lie upstream and within promoters [53,54,55].
The design of synthetic eukaryotic promoters has traditionally implemented a common architecture, where a strong transcriptional initiation region is cloned downstream of orthogonal DNA binding operator sequences and the latter serve as landing pads for synthetic transcription factors [23]. The engineered transcription factors have typically consisted of DNA binding proteins (i.e., prokaryotic DNA binding proteins like TetR, LacI, LexA and PhIF [56,57,58]) fused to well characterized transcriptional activation domain like VP64. With the advent of programmable DNA binding proteins like zinc finger proteins and TALEs, the repertoire of synthetic promoters greatly increased [23, 24, 59, 60]. That said, each new synthetic promoter still requires the construction and characterization of its own unique transcription factor [23, 60, 61].
These bottlenecks can be circumvented by the use of the highly programmable RNA-guided DNA binding protein dCas9 [26]. The dCas9 RNP fused to transcription activation domains such as VP64 has been used for the upregulation of endogenous genes in a wide variety of eukaryotic species like yeast, mammalian cells and plants [16, 25, 26, 62]. Here, we have used adapted this ‘universal’ transcription factor to control the expression of synthetic and orthogonal promoters without the need of addition of any other factors. Using our modular framework, we were able to quickly design and characterize a panel of mutually orthogonal promoters that could drive the production of a variety of outputs, singly and in parallel, including different fluorescent proteins (GFP, BFP, RFP and YFP) and luciferase.
The activities of dCas9 based transcription factors can potentially be controlled by simply regulating the expression of their corresponding gRNAs [16, 17], enabling the coupling of natural and synthetic transcription units, and thus natural and overlaid metabolic responses. Here we have effectively used this strategy to couple ethylene sensing (via known EIN3 binding sites) to synthetic (pATF) promoters. Moreover, by changing the number and arrangement of gRNA binding sites synthetic promoters with different levels of activation can be generated, providing further opportunities for design [63]. While it has been previously shown that a panel of minimal plant promoters can be used with natural DNA binding sequences for modulating promoter strengths [20], the addition of completely artificial, synthetic promoters as control elements should create opportunities for increasing the specificity and strengths of engineered promoters.
Since our strategy for designing synthetic promoters is generalizable it is likely that even more complex circuits can be built by simply incorporating other transcription factor binding sites, or by changing the regulatory ‘headpiece’ on the dCas9 element (for example, to a repressor) [64,65,66]. In particular, the activity of synthetic promoters depends on the strength of the activation domains fused to dCas9. In this work we have utilized the highly characterized activation domain – VP64. More recently, potent dCas9-based synthetic transcription factors have been described that exhibit even higher activity than dCas9:VP64 [67, 68]. The modular nature of the OCS architecture described in this work complements the incorporation of other activation domains into dCas9-based transcription factors and would further augment the activity of OCS circuitry.
The stabilities of genetic circuitry in plants can be greatly modified by silencing and recombination, amongst other mechanisms [40, 41, 43]. In this regard, the artificial promoter elements that we generate can potentially be crafted to avoid repetition [20], and thus to better avoid silencing and recombination. As viable artificial promoter sequences continue to accumulate, they can be compared and contrasted to identify those that are least vulnerable to modification over time. While in this work we have focused on the use of highly active 35S terminator, the construction of more stable circuits may require the use of new terminator signals. The facile addition of new parts to the standardize toolkit architecture, particularly terminators, will further increase opportunities to avoid repetition in ways that again go well beyond what is possible by relying on just a few well-characterized endogenous elements alone.
The implementation of orthogonal control systems in plants can be used to limit cross-talk between natural and overlaid regulatory elements, allowing more precise response to a variety of inputs, from VOCs to hormones to temperature, water, and nutrients. The use of orthogonal control systems to enable more precise responses to pathogenesis is especially intriguing given the presence of R genes that are specifically responsive to individual pathogens (effector triggered immunity, ETI) [69]. The architecture we have developed is fully generalizable, and can potentially be expanded to non-model plants and other eukaryotic species such as yeast and mammalian cells by the use of appropriate transcription initiation regions under the control of similar gRNA sequences binding sites [70].
Conclusion
The design of synthetic promoters based on gRNAs and CRISPR-based transcription factor (dCas9:VP64) is highly generalizable to large tracts of sequence space, allowing Orthogonal Control Systems of increasing complexity to potentially be generated at will. The ability to tie in several different basal features of plant molecular biology (Pol II and Pol III promoters, ethylene regulation) to the OCS demonstrates multiple opportunities for engineering at the system level. Moreover, given the fungibility of the core 35S CaMV promoter elements, the derived synthetic promoters can potentially be utilized across a variety of plant species.
Materials and methods
Plasmid design and construction
The plant expression vector was generated using the plasmid pICH86966 (Addgene#48075) as the backbone. The lacZ expression cassette was replaced with the GFP dropout sequence (Additional file 1: Table S2) to make the plasmid compatible with YTK architecture design. All parts described in Additional file 1: Table S1, were cloned into the backbone pYTK001 (Addgene #65108). For the individual transcriptional units, the backbone used was pYTK095 (Addgene #65202) along with the appropriate connector sequences described in Additional file 1: Table S3. For the design of orthogonal gRNAs, random 20-mers were generated that had a GC content of ~ 50%, and that were at least 5 nucleotides away from all sequences in the Nicotiana and Arabidopsis genomes. All oligonucleotides and gblocks were obtained from Integrated DNA Technologies (IDT) unless otherwise stated.
For the construction of each genetic element namely promoters, coding sequences and terminators, first they were checked for restriction sites for the following enzymes – BsmBI, BsaI, NotI and DraIII. The restriction sites in the coding sequences were removed by the use of synonymous codons while the other elements did not contain any of these restriction sites. The complete list of parts and constructs are provided in Additional file 1: Table S1. The part plasmids were cloned into a common vector where each genetic element is flanked by Bsa1 restriction sites followed by appropriate overhangs (Additional file 1: Table S1). For the assembly of both single TU or multi-TU, the following procedure was used: 10 fmol of backbone plasmid and 20 fmol of parts/TUs were used in a 10uL reaction with 1ul of 10 × T4 ligase buffer along with 100 units of BsaI-v2 (single TU) or Esp3I (multi-TU or parts) and 100 units of T7 DNA ligase. The cycling protocol used is: 24 cycles of 3 min at 37 °C (for digestion) and 5 min at 16 °C (for ligation) followed by a final digestion step at 37 °C for 30 min and the enzymes were heat inactivated 80 °C for 20 min. All constructs were transformed into DH10B cells, grown at 37 °C using standard chemical transformation procedures. The colonies that lack fluorescence were inoculated and plasmids were extracted using Qiagen Miniprep kit according to the manufacturer’s instructions Plasmids were maintained as the following antibiotics kanamycin (50 μg/mL), chloramphenicol (34 μg/mL) and carbenicillin (100 μg/mL) wherever required. The plasmids were sequence verified by Sanger sequencing (UT Austin Genomic Sequencing and Analysis Facility). The correct constructs were then transformed into Agrobacterium tumefaciens strain GV3101 (resistant to Gentamycin and Rifampicin) and used either for transient expression in Nicotiana benthamiana or to generate stable lines in Arabidopsis thaliana. The following enzymes were used for the assemblies – BsaI-v2 (NEB #R3733S), Esp3I (NEB #R0734S) and T7 DNA ligase (NEB #M0318S).
Plant material, bacterial infiltration
Nicotiana benthamiana and Arabidopsis thaliana plants were grown in soil at 22 °C, and 16 h light period. For transient expression, three weeks old plants were syringe-infiltrated with Agrobacterium tumefaciens strain GV3101 (OD600 = 0.5) and leaves were imaged under Olympus BX53 Digital Fluorescence Microscope or harvested for RNA and/or protein analysis. To create stable transformation in Arabidopsis, floral dip method [71] was used. T1 plants were selected on half MS Kanamycin (50 μg/ml) plates and the selected T1 plants were analyzed using an Olympus BX53 Digital Fluorescence Microscope and a NightOwl imager for YFP expression and luciferase expression, respectively. For circuits that constitutively expressed YFP (OCS1-1) and luciferase (OCS4-1) no other obvious phenotypic differences were observed across numerous individual plants.
RNA extraction and qRT-PCR
RNA was extracted using TRIzol reagent (Ambion). 1 μg total RNA was used to synthesize cDNA. After DNaseI treatment to remove any DNA contamination, random primer mix (NEB #S1330S) and M-MLV Reverse transcriptase (Invitrogen #28,025–013) were used for first strand synthesis. qRT-PCR was used to quantify the RNA prepared from transient expression experiments. AzuraQuant qPCR Master Mix (Azura Genomics) was used with initial incubation at 95 °C for 2 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Level of target RNA was calculated from the difference of threshold cycle (Ct) values between reference (5S rRNA) and target gene using at least three independent replicates.
ACC treatment
To check the induction of reporter in response to ACC in the plasmids containing pEBS::YFP/RFP/BFP, Nicotiana benthamiana leaves were infiltrated with Agrobacterium; after three days post infiltration, leaf discs were cut using cork borer and incubated in either 0 μM or 10 μM ACC for four hours. Fluorescence microscopy was used to check YFP expression after induction.
Fluorescence and Luminescence imaging
Fluorescence microscope images after Agrobacterium mediated transient expression of YFP, BFP, RFP and GFP in Nicotiana benthamiana leaves were taken using an Olympus BX53 Digital Fluorescence Microscope. For this purpose, leaf discs were cut using cork borer from the area which was infiltrated. Images were taken using either 10X objective lens using the default filters for YFP (500/535 nm), BFP (385/448 nm), and RFP (560/630 nm). The UV filter (350/460 nm) was used to take GFP images. The exposure and gain setting were kept constant for each filter within each experiment to compare multiple leaf discs (3 to 6). In all the experiments a leaf disc from a leaf which was not infiltrated with Agrobacterium was used as a negative control in order to account for background fluorescence. All experiments were performed at least three times independently as indicated in the Results.
Expression of luciferase was detected using NightOwl II LB 983 in vivo imaging system (https://www.berthold.com/en/bioanalytic/products/in-vivo-imaging-systems/nightowl-lb983/). Leaves/plants were sprayed with 100 μM D-luciferin, Potassium salt (GoldBio #LUCK-300). After 5 min of incubation, images were taken in the NightOwl II LB 983. Images were captured with a backlit NightOWL LB 983 NC 100 CCD camera. Photons emitted from luciferase were collected and integrated for a 2 min period. A pseudocolor luminescent image from blue (least intense) to red (most intense), representing the distribution of the detected photons emitted from active luciferase was generated using Indigo software (Berthold Technologies).
Western blot
Total protein was extracted using urea-based denaturing buffer (100 mM NaH2PO4, 8 M urea, and 10 mM Tris–HCl, pH 8.0) and used for immunoblot analysis to check the expression. The proteins were fractionated by 8% SDS-PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane using a transfer apparatus according to the manufacturer’s protocols (Bio-Rad). The membrane was treated with 5% nonfat milk in PBS-T for 10 min for blocking, and then incubated with Cas9 antibody (Santa cruz, 7A9-3A3, 1:500) at 4 °C for overnight. After incubation, the membrane was washed three times for 5 min and incubated with horseradish peroxidase-conjugated anti-mouse (1:10,000) for 2 h. The Blot was washed with PBS-T three times and detected with the ECL system (Thermo scientific, lot# SE251206).
Availability of data and materials
The datasets during and/or analysed during the current study available from the corresponding author on reasonable request.
References
Tolle F, Stucheli P, Fussenegger M. Genetic circuitry for personalized human cell therapy. Curr Opin Biotechnol. 2019;59:31–8.
Scheller L, Fussenegger M. From synthetic biology to human therapy: engineered mammalian cells. Curr Opin Biotechnol. 2019;58:108–16.
Sedlmayer F, Aubel D, Fussenegger M. Synthetic gene circuits for the detection, elimination and prevention of disease. Nat Biomed Eng. 2018;2(6):399–415.
Riglar DT, Silver PA. Engineering bacteria for diagnostic and therapeutic applications. Nat Rev Microbiol. 2018;16(4):214–25.
Kassaw TK, Donayre-Torres AJ, Antunes MS, Morey KJ, Medford JI. Engineering synthetic regulatory circuits in plants. Plant Sci. 2018;273:13–22.
de Lange O, Klavins E, Nemhauser J. Synthetic genetic circuits in crop plants. Curr Opin Biotechnol. 2018;49:16–22.
Nielsen AA, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, et al. Genetic circuit design automation. Science (New York, NY). 2016;352(6281):aac7341.
Medford JI, Prasad A. Towards programmable plant genetic circuits. Plant J. 2016;87(1):139–48.
Fonseca JP, Bonny AR, Kumar GR, Ng AH, Town J, Wu QC, et al. A toolkit for rapid modular construction of biological circuits in mammalian cells. ACS Synth Biol. 2019;8(11):2593–606.
Brophy JA, Voigt CA. Principles of genetic circuit design. Nat Methods. 2014;11(5):508–20.
Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai QA, et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods. 2013;10(4):354–60.
Weinberg BH, Pham NTH, Caraballo LD, Lozanoski T, Engel A, Bhatia S, et al. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat Biotechnol. 2017;35(5):453–62.
Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature. 2005;434(7037):1130–4.
Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403(6767):339–42.
Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403(6767):335–8.
Kim H, Bojar D, Fussenegger M. A CRISPR/Cas9-based central processing unit to program complex logic computation in human cells. Proc Natl Acad Sci USA. 2019;116(15):7214–9.
Gander MW, Vrana JD, Voje WE, Carothers JM, Klavins E. Digital logic circuits in yeast with CRISPR-dCas9 NOR gates. Nat Commun. 2017;8:15459.
Weber W, Fussenegger M. Engineering of synthetic mammalian gene networks. Chem Biol. 2009;16(3):287–97.
Kramer BP, Fussenegger M. Hysteresis in a synthetic mammalian gene network. Proc Natl Acad Sci USA. 2005;102(27):9517–22.
Belcher MS, Vuu KM, Zhou A, Mansoori N, Agosto Ramos A, Thompson MG, et al. Design of orthogonal regulatory systems for modulating gene expression in plants. Nat Chem Biol. 2020;16(8):857–65.
Schaumberg KA, Antunes MS, Kassaw TK, Xu W, Zalewski CS, Medford JI, et al. Quantitative characterization of genetic parts and circuits for plant synthetic biology. Nat Methods. 2016;13(1):94–100.
Brückner K, Schäfer P, Weber E, Grützner R, Marillonnet S, Tissier A. A library of synthetic transcription activator-like effector-activated promoters for coordinated orthogonal gene expression in plants. Plant J. 2015;82(4):707–16.
Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, Joung JK, et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 2012;150(3):647–58.
Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, et al. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol. 2003;21(3):275–80.
Li Z, Zhang D, **ong X, Yan B, **e W, Sheen J, et al. A potent Cas9-derived gene activator for plant and mammalian cells. Nat Plants. 2017;3(12):930–6.
Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–6.
Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, et al. Comparison of Cas9 activators in multiple species. Nat Methods. 2016;13(7):563–7.
Lee ME, DeLoache WC, Cervantes B, Dueber JE. A highly characterized yeast toolkit for modular multipart assembly. ACS Synth Biol. 2015;4(9):975–86.
Engler C, Youles M, Gruetzner R, Ehnert TM, Werner S, Jones JD, et al. A golden gate modular cloning toolbox for plants. ACS Synth Biol. 2014;3(11):839–43.
Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS One. 2011;6(2):e16765.
Banta LM, Montenegro M. Agrobacterium and Plant Biotechnology. In: Tzfira T, Citovsky V, editors. Agrobacterium: From Biology to Biotechnology. New York, NY: Springer New York; 2008. p. 73–147.
Berg RH, Beachy RN. Fluorescent protein applications in plants. Methods Cell Biol. 2008;85:153–77.
Benfey PN, Chua NH. The Cauliflower Mosaic Virus 35S Promoter: Combinatorial Regulation of Transcription in Plants. Science (New York, NY). 1990;250(4983):959–66.
Odell JT, Nagy F, Chua NH. Identification of DNA sequences required for activity of the cauliflower mosaic virus 35S promoter. Nature. 1985;313(6005):810–2.
Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37(1):465–87.
Tindall AJ, Waller J, Greenwood M, Gould PD, Hartwell J, Hall A. A comparison of high-throughput techniques for assaying circadian rhythms in plants. Plant Methods. 2015;11(1):32.
**ong TC, Sanchez F, Briat J-F, Gaymard F, Dubos C. Spatio-Temporal Imaging of Promoter Activity in Intact Plant Tissues. In: Hehl R, editor. Plant Synthetic Promoters: Methods and Protocols. New York, NY: Springer New York; 2016. p. 103-10.
Xu G, Greene GH, Yoo H, Liu L, Marqués J, Motley J, et al. Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature. 2017;545(7655):487–90.
Zhou M, Wang W, Karapetyan S, Mwimba M, Marqués J, Buchler NE, et al. Redox rhythm reinforces the circadian clock to gate immune response. Nature. 2015;523:472.
Vaillant I, Schubert I, Tourmente S, Mathieu O. MOM1 mediates DNA-methylation-independent silencing of repetitive sequences in Arabidopsis. EMBO Rep. 2006;7(12):1273–8.
Matzke MA, Mette MF, Matzke AJ. Transgene silencing by the host genome defense: implications for the evolution of epigenetic control mechanisms in plants and vertebrates. Plant Mol Biol. 2000;43(2–3):401–15.
Ni M, Cui D, Einstein J, Narasimhulu S, Vergara CE, Gelvin SB. Strength and tissue specificity of chimeric promoters derived from the octopine and mannopine synthase genes. Plant J. 1995;7(4):661–76.
Matzke MA, Primig M, Trnovsky J, Matzke AJ. Reversible methylation and inactivation of marker genes in sequentially transformed tobacco plants. Embo j. 1989;8(3):643–9.
Cardinale S, Arkin AP. Contextualizing context for synthetic biology – identifying causes of failure of synthetic biological systems. Biotechnol J. 2012;7(7):856–66.
Kang H-G, Fang Y, Singh KB. A glucocorticoid-inducible transcription system causes severe growth defects in Arabidopsis and induces defense-related genes. Plant J. 1999;20(1):127–33.
Amirsadeghi S, McDonald AE, Vanlerberghe GC. A glucocorticoid-inducible gene expression system can cause growth defects in tobacco. Planta. 2007;226(2):453–63.
Lowder LG, Paul JW 3rd, Qi Y. Multiplexed Transcriptional Activation or Repression in Plants Using CRISPR-dCas9-Based Systems. Methods Mol Biol. 2017;1629:167–84.
Bayer TS, Smolke CD. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat Biotechnol. 2005;23(3):337–43.
Kotula JW, Kerns SJ, Shaket LA, Siraj L, Collins JJ, Way JC, et al. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc Natl Acad Sci. 2014;111(13):4838.
Jacobs JZ, Ciccaglione KM, Tournier V, Zaratiegui M. Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun. 2014;5(1):5344.
Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell. 2014;54(4):698–710.
Cruz AB, Bianchetti RE, Alves FRR, Purgatto E, Peres LEP, Rossi M, et al. Light, Ethylene and Auxin Signaling Interaction Regulates Carotenoid Biosynthesis During Tomato Fruit Ripening. Front Plant Sci. 2018;9:1370.
Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68.
Ellwood K, Huang W, Johnson R, Carey M. Multiple layers of cooperativity regulate enhanceosome-responsive RNA polymerase II transcription complex assembly. Mol Cell Biol. 1999;19(4):2613–23.
Chen L. Combinatorial gene regulation by eukaryotic transcription factors. Curr Opin Struct Biol. 1999;9(1):48–55.
Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci USA. 2000;97(14):7963–8.
Bellí G, Garí E, Piedrafita L, Aldea M, Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 1998;26(4):942–7.
Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89(12):5547–51.
Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F, et al. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods. 2013;10(3):239–42.
Li Y, Moore R, Guinn M, Bleris L. Transcription activator-like effector hybrids for conditional control and rewiring of chromosomal transgene expression. Sci Rep. 2012;2:897.
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82.
Lowder LG, Zhang D, Baltes NJ, Paul JW 3rd, Tang X, Zheng X, et al. A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol. 2015;169(2):971–85.
Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol. 2019;37(6):657–66.
Pandelakis M, Delgado E, Ebrahimkhani MR. CRISPR-based synthetic transcription factors in vivo: the future of therapeutic cellular programming. Cell Syst. 2020;10(1):1–14.
Yeo NC, Chavez A, Lance-Byrne A, Chan Y, Menn D, Milanova D, et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods. 2018;15(8):611–6.
Kwon DY, Zhao YT, Lamonica JM, Zhou Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat Commun. 2017;8:15315.
**ong X, Liang J, Li Z, Gong BQ, Li JF. Multiplex and optimization of dCas9-TV-mediated gene activation in plants. J Integr Plant Biol. 2021;63(4):634–45.
Pan C, Wu X, Markel K, Malzahn AA, Kundagrami N, Sretenovic S, et al. CRISPR-Act30 for highly efficient multiplexed gene activation in plants. Nat Plants. 2021;7(7):942–53.
Gonzalez TL, Liang Y, Nguyen BN, Staskawicz BJ, Loqué D, Hammond MC. Tight regulation of plant immune responses by combining promoter and suicide exon elements. Nucleic Acids Res. 2015;43(14):7152–61.
Farzadfard F, Perli SD, Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol. 2013;2(10):604–13.
Zhang X, Henriques R, Lin S-S, Niu Q-W, Chua N-H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat Protoc. 2006;1(2):641–6.
Acknowledgements
We would also like to thank the Qiao lab (UT Austin) for providing details regarding the ethylene induction of the OCS constructs.
Funding
This work was supported by the Defense Advanced Research Projects Agency (DARPA) agreement HR00111820048 to AE and SS. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. This work was also supported by Welch Foundation grant (F-1654) to ADE.
Author information
Authors and Affiliations
Contributions
SK, SS and AE conceived of the project. SK designed the framework and the basic elements of OCS with input from EG, JG and SS. SK and YB assembled all constructs. YB, NR and JK performed all the testing in Nicotiana with input from SS. All authors contributed with the preparation of figures. SK, YB, JK, SS and AE wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1
: Workflow describing the assembly of single and multiple transcriptional units (TUs) in a plant expression vector; Fig S2: Western blot to analyze the expression of dCas9:VP64 in OCS constructs – OCS1-1 and OCS 1–5. Table S1: List of all genetic parts used for the construction of OCS constructs. Table S2: List of all OCS constructs. Table S3: List of all Addgene plasmids used in this work
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kar, S., Bordiya, Y., Rodriguez, N. et al. Orthogonal control of gene expression in plants using synthetic promoters and CRISPR-based transcription factors. Plant Methods 18, 42 (2022). https://doi.org/10.1186/s13007-022-00867-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-022-00867-1