
Abstract
The Flaviviridae virus family members cause severe human diseases and are responsible for considerable mortality and morbidity worldwide. Therefore, researchers have conducted genetic screens to enhance insight into viral dependency and develop potential anti-viral strategies to treat and prevent these infections. The host factors identified by the clustered regularly interspaced short palindromic repeats (CRISPR) system can be potential targets for drug development. Meanwhile, CRISPR technology can be efficiently used to treat viral diseases as it targets both DNA and RNA. This paper discusses the host factors related to the life cycle of viruses of this family that were recently discovered using the CRISPR system. It also explores the role of immune factors and recent advances in gene editing in treating flavivirus-related diseases. The ever-increasing advancements of this technology may promise new therapeutic approaches with unique capabilities, surpassing the traditional methods of drug production and treatment.
Similar content being viewed by others
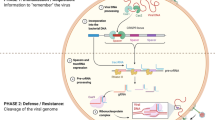

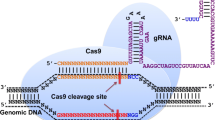
Introduction
The Flaviviridae family encompasses a large group of single-stranded, positive-sense RNA viruses. Four genera belong to this family: Flavivirus, Pestivirus, Hepacivirus, and Pegivirus. Some members of the Hepacivirus and Flavivirus genera are responsible for several important human diseases [1]. Hepatitis C virus (HCV) belongs to the Hepacivirus genus, which differs in many aspects compared to the members of the Flavivirus genus, including the transmission route or the course of infection [2]. The main transmission path for most flaviviruses is through arthropod vectors and includes important pathogens such as the Zika virus (ZIKV) [3]. Zika virus infections in pregnant women have been associated with congenital microcephaly and other developmental defects in infants. The aforementioned traits of Zika virus has attracted the attention of the medical community worldwide [4,5,6]. Dengue virus (DENV), which causes approximately 100 million symptomatic infections annually, is another cause of infectious diseases inflicted by flaviviruses [7]. Yellow fever virus (YFV) is another member of the Flavivirus genus and is known to be a cause of hemorrhagic fever. It remains prevalent in sub-Saharan Africa and South America, in spite of the availability of a highly effective live-attenuated vaccine against it [8, 9]. Among people infected with West Nile virus (WNV), only about 20% present the symptoms of West Nile fever (WNF). Less than 1% of the infected individuals develop a neuroinvasive disease characterized by encephalitis, meningitis, and flaccid paralysis [10,11,12]. So far, no specific or potent antiviral treatments are available against ZIKV, DENV, and WNV infections. Outbreaks still occur despite licensed vaccines against several members of the Flaviviridae family, including DENV, YFV, Japanese encephalitis virus (JEV), and Tick-borne encephalitis virus (TBEV), emphasizing the challenges and flaws in implementing effective vaccination programs [13]. Viruses are obligate pathogens, dependent on their host to complete their replication cycle. Viruses utilize cellular receptors to enter the host and hijack cellular functions and pathways to replicate, assemble and release new virus particles; hence, identifying the cellular factors that promote or restrict virus replication will reveal the fundamental characteristics of host-virus interaction. This, in turn, could lead to the development of target-specific antiviral drugs in the future [14]. Genomic approaches are increasingly being utilized to identify viral pathogenesis mechanisms and study host-viral interactions. Several genetic screening technologies, such as RNA interference (RNAi), haploid embryonic stem cells, and clustered regularly interspaced short palindromic repeats (CRISPR), have proven to be powerful means for examining viral lifecycles [15]. However, the CRISPR/Cas technology as an efficient tool for genomic engineering has overcome the limitations of other competing technologies. Furthermore, this system has been engineered to effectively induce knockout mutations in a wide range of cell types. The expansion of CRISPR/Cas9 screening libraries allows all known genes from any species to be targeted, including a pool of guide RNAs to target a vast variety of genes. Either way, gene knockouts or the activation of gene expression can be achieved [16, 17]. The present review aims to elucidate the basic principles and types of different CRISPR screens, and their use in novel anti-viral approaches. Therefore, we have also provided a comprehensive overview of the recent discoveries about virus-host interactions which have been achieved using CRISPR screens. Lastly, we have described the currently available CRISPR-Cas antiviral strategies against the Flaviviridae family as one of the main groups of lethal viral infections.
CRISPR/Cas system
CRISPR/Cas system is an adaptive immune mechanism protecting the bacteria against invading viruses and plasmids [18, 19]. Bacterial CRISPR loci consist of a Cas operon and a repeat-spacer array. This defensive process can be divided into three stages. The acquisition step involves the integration of foreign nucleic acids into a CRISPR array as new CRISPR spacers separated by repeat sequences found adjacent to the CRISPR-associated (Cas) genes, which encode Cas ribonucleases. This step creates a memory of the foreign genetic components [20]. In the second step (expression), the CRISPR array must be transcribed into a pre-CRISPR RNA transcript (pre-crRNA), then processed. The outcome of this step at this stage is finally a mature crRNA [21, 22]. Additionally, a transactivating RNA (tracrRNA) is also encoded by the CRISPR locus, which has complementarity properties to the repeat areas of crRNA transcripts [21]. Subsequently, in the third stage (i.e. interference), through the binding of complementary repeat region sequences, the crRNA-tracrRNA hybrid is formed. Finally, Cas nuclease is guided to the complementary DNA sequences using this RNA hybrid, targeting and cleaving the nucleic acids derived from the invading viruses and other genetic elements [23].
The CRISPR/Cas systems are divided into two categories according to their effector molecules: multi-subunit effector molecules in Class 1 and a single effector molecule in Class 2. The first class can be subdivided into three types (I, III, and IV), and Class 2 consists of types II, V, and VI [24, 25]. Specifically, the CRISPR/Cas9 system, which belongs to Class 2 (type II), involves the association of crRNA with a single unit of Cas protein (Cas9) for its function [26]. This system, which can potentially edit any gene or genomic region, is widely used, including in virology. The functional complex comprises Cas9 and a single-guide RNA (sgRNA); TracrRNA and crRNA can be fused into a sgRNA. According to recent studies, class 2 type VI CRISPR effector Cas13 can efficiently target and cleave RNA instead of DNA in different model systems, including mammalian cells [27, 28]. Thus, the CRISPR-Cas13 system offers the potential to detect RNA viruses and treat RNA virus infections. Cas13 proteins have been classified into several types, each containing two higher eukaryotes and prokaryotes’ nucleotide (HEPN)-binding domains necessary for RNA degradation [29,30,67]; and second, components of the ER membrane complex (EMC) [66]. The mosquito-borne flaviviruses, including DENV, ZIKV, and WNV, were found to be dependent on the EMC for the expression of their viral polyproteins. Specifically, the EMC engages with transmembrane domains (TMDs) in NS4A and NS4B during the translation process to ensure accurate topology, correct folding, and stable expression. Ngo et al. observed a notable decrease in viral RNA, especially for WNV, ZIKV, and DENV, but not for HCV, after EMC4 knockout [51]. According to a study by Ma et al., the knockout of several genes, including EMC2, EMC3, SEL1L, DERL2, UBE2G2, UBE2J1, and HRD1, inhibited WNV-induced cell death but did not affect WNV replication. These seven genes of the ERAD pathway connect WNV replication to downstream cell death pathway(s) [57]. Neuronal cell death is one of the most common causes of death due to WNV infection, and these proteins might also serve as new therapeutic targets. Furthermore, another study found that ZIKV, DENV, and YFV strongly required EMC for replication in the early stages of infection [54]. The results of these studies show distinct roles for EMC in the flavivirus infection cycle. The results first emphasize the effective role of EMC in replicating DENV, ZIKV, and YFV viruses. They then describe its function, which is required for WNV-induced cytopathic role (cytopathicity). However, the exact mechanism through which EMC acts in flavivirus infections should be examined more broadly.
TMEM41B and VMP1 proteins
Transmembrane protein 41B (TMEM41B) is a multi-spanning membrane protein found in the endoplasmic reticulum (ER). TMEM41B has similar roles to vacuole membrane protein 1 (VMP1), such as autophagy and lipid mobilization [68,69,70]. The CRISPR/Cas9 screens have recently revealed that TMEM41B is a pan-flavivirus host factor; it has also been identified that in addition to ZIKV, YFV, HCV, WNV, and DENV-2, several other members of the Flaviviridae family require TMEM41B for causing infection. TMEM41B and VMP1 might also be involved in remodeling cell membranes through their association with flavivirus proteins. Indeed, flaviviruses may hijack these proteins for their ability to remodel host cell membranes that are required to form viral replication compartments in the ER. In particular, Hoffmann et al. have suggested that together with NS4A and NS4B, TMEM41B is recruited at the ER membrane where replication complexes are formed, then TME41B reduces the local free energy imposed by the NS4A- and NS4B-induced membrane curvatures (Fig. 2) [55].

Flavivirus replication cycle and the role of TMEM41B as an ER remodeling protein. A: wild‐type cells; B: TME41B KO cells
DPMS complex
Utilizing a genome-wide CRISPR/Cas9 screen, Athena et al. identified two resident subunits of the endoplasmic reticulum incorporating dolichol-phosphate mannose synthase (DPMS) complex, DPM1 and DPM3, and the role they play in DENV infection. They discovered that DPM1 or DPM 3 are required for the efficient infection by all the DENV serotypes. Moreover, ZIKV and YFV 17D infection was remarkably inhibited in cells lacking DPM1 or DPM3. According to this study, DENV requires DPMS to regulate viral RNA replication and proper glycosylation of the viral proteins E, prM, and NS1 [52].
RACK1
The Receptor for Activated Protein C Kinase 1 (RACK1) is a core component of the 40S ribosomal subunit and has a significant role in several aspects of cellular functions [71, 72]. Recently, according to a CRISPR/Cas9 KO screen, RACK1 was identified as a novel host factor that seems to be required for ZIKV replication. The function of this factor is crucial for viral RNA genome replication. In particular, NS1 is essential for the biogenesis of viral replication factories known as ‘vesicle packets’ (VPs), and RACK1, through interaction with NS1 within the ER lumen, playing a key role in the construction of replication organelles early in the virus lifecycle. As a result, RACK1 depletion leads to changes in morphology and decreases the frequency of VPs [56]. In addition, another study examined the function of RACK1 during the life cycle of DENV. RACK1, in association with Vigilin and SERBP1 factors which interact with DENV viral RNA, forms a ternary complex mediating viral replication [53].
TRIM26
The host factors involved in HCV replication are also of tremendous importance. Liang et al., using a CRISPR/Cas9 screen, showed that TRIM26, an E3 ubiquitination ligase, is a critical host factor for HCV (Fig. 3). They revealed that TRIM26 interacts with the NS5B protein and thus mediates its K27-linked ubiquitination at residue K51, increasing NS5B-NS5A interaction. The knockout of TRIM26 significantly diminished HCV replication, but did not terminate it entirely. In contrast, TRIM26 seems to play a virus-specific role in HCV replication since it is not involved in DENV and ZIKV life cycles [61].

The role of TRIM26 in promoting HCV genome replication. A: wild‐type cells; B: TRIM26 knockout cells
ELAVL1, RFK, and FLAD1
One of the RNA-binding proteins involved in mRNA stabilization is ELAVL1; since it has been observed that HCV RNA replication in the ELAVL1-knockout cells was significantly reduced. This suggests that ELAVL1 is crucial for the HCV RNA replication. HCV screens have also contributed to the discovery of the role of the enzymes involved in the HCV replication. For example, riboflavin kinase (RFK) and FAD synthase (FLAD1) knockout cells, two enzymes involved in the conversion of riboflavin (vitamin B2) to FAD, were resistant to HCV replication. This is while FAD rescued HCV replication in these knockout cells; therefore, a link between intracellular FAD levels and HCV RNA replication was discovered. Hence, Lumiflavin, which is an inhibitor of the cellular uptake of riboflavin, can inhibit viral RNA replication [14].
Polyprotein processing and viral translation
The signal peptidase complex (SPC) is a membrane complex in the endoplasmic reticulum, where it cleaves signal peptides (SPs) from the N-termini of the secretory and membrane proteins. A subset of signal peptidase complex (SPCS) proteins is required for efficient cleavage of flavivirus structural proteins (prM and E) and the secretion of viral particles; based on the CRISPR/Cas9 screen, Knockout of this factor indicated intense defects in the polyprotein cleavage of several Flaviviridae family members [58]. Another known factor is the translocon-associated protein (TRAP) complex (containing subunits SSR1, SSR2, and SSR3), which facilitates the translocation of proteins across the endoplasmic reticulum membrane [73]. The TRAP complex plays a crucial role in DENV-2, ZIKV, and YFV RNA replication [14]. A recent study has shown that host factors SBDS and SPATA5, which are involved in the formation of ribosomes, are required for the synthesis of viral proteins. Losing these 60S ribosome biogenesis proteins leads to a decrease in the viral replication of flaviviruses and several other viral families. The study specifically found that the loss of function of these two proteins, which are essential for the formation of the 60S ribosome part, causes defects in the processing of rRNAs and the assembly of ribosomes [74].
Anti-viral immunity
Host antiviral restriction factors
Although many host proteins are hijacked or disrupted by viral infections, various studies have shown that several host proteins operate with antiviral properties acting against viral infection. In this regard, a gain-of-function screen has introduced TMEM120A as a host restricting factor against ZIKV infection. TMEM120A is a transmembrane protein localized on the plasma membrane, nuclear membrane, and endoplasmic reticulum [75,76,77]. TMEM120A displays an antiviral function through interaction with STING; it promotes STING translocation from the endoplasmic reticulum to the ER-Golgi intermediate compartment (ERGIC), leading to the activation of TBK1 and phosphorylation of the transcription factor IRF3. Subsequently, it results in the activation of type-I interferon (IFN) expression (Fig. 4A) [77]. On the other hand, CRISPR screens have discovered interferon responses to flaviviruses. The IFN response can target viral replication at the ER. For instance, IFI6 (encoding Interferon alpha-inducible protein 6) and HSPA5 (encoding endoplasmic reticulum chaperone BiP) are two genes involved in this antiviral mechanism. IFI6, localized to the ER and stabilized by its interaction with BiP, prevents the formation of virus-induced ER membrane invaginations, hence suppressing the viral life cycle (Fig. 4B) [49]. Moreover, a CRISPR activation screen recognized that IFI6 and other ISGs, including Interferon Lambda 2 (IFN-λ2), can rescue cells from ZIKV infection. On the other hand, it has recently been found that ZIKV can evade the BiP/HSPA5 pathway by down regulating their expression (78). The aforementioned point alongside the other mostly controversial roles suggested about the mechanisms of this pathway, necessitates further studies to be conducted to clarify whether the BiP/HSPA5 can lead to novel antiviral therapies. Collectively, these studies show the potential of various CRISPR screening strategies in recognizing host factors that either facilitate or inhibit viral replication [79].

A Schematic of the antiviral function of TMEM120A. B The antiviral role of IFI6 that inhibits the formation of a replication organelle through interaction with BiP
Regulation of interferon signaling pathways
CRISPR screening allows researchers to identify genes regulating interferon (IFN) responses to flavivirus infections. According to the investigations, ZIKV causes proliferation loss and cell death in human neural progenitors (NPs) during early cortical development, resulting in fetal brain abnormality. In this regard, a study identified host genes in human NPs associated with ZIKV infection, such as host factors involved in the interferon activity. The results revealed that knocking out IFN pathway regulators in NPs, including ISG15 and SOCS3, reduced the infectivity of ZIKV. Therefore, ZIKV seems to evade the host antiviral defense by relying on negative regulators of the IFN pathway [80].
Viral RNAs are identified by RIG-I-like receptors (RLRs), including RIG-I and MDA5, leading to the activation of RIG-I/MAVS, a significant immune pathway [81]. Therefore, inactivating RIG-I impairs the response against these pathogens. CRISPR-KO demonstrated that RIG-I, but not MDA5, was the major sensor for the recognition of ZIKV RNA in A549 cells. When RIG-I recognizes ZIKV infection, it ultimately leads to the induction of type I IFNs and ISGs. The lack of RIG-I causes ISG expression to diminish in the ZIKV-infected cells, increasing the viral replication and apoptosis [82]. RNase L, an antiviral enzyme with several functions such as the degradation of viral and cellular RNAs, inhibiting protein synthesis, and restricting the replication and spread of various viruses, is activated by the 2′,5′ -oligoadenylates after the infection with RNA viruses. It should be noted that different oligoadenylate synthetases (OAS), OAS1, OAS2, and OAS3, synthesize 2′,5′-oligoadenylates. Using CRISPR-KO, it was uncovered that in this process, OAS3, but not OAS1 or OAS2, is a major factor involved in 2′,5′-oligoadenylate synthesis for RNAse L activation. This study also revealed that in RNase L- and OAS3-KO cells, the replication of four viruses, including WNV, was increased. According to these results, OAS3 may serve as an antiviral target, while OAS1 and OAS2 may have alternative functions [83]. Studies have been conducted to confirm and characterize the main components of IFN pathways using the CRISPR system. A study explored the function of STAT1 and STAT2 in inhibiting HCV replication through IFN-α and IFN-λ. STAT1 and STAT2 play a role in the early induction of ISGs in response to IFN-α in Huh-7.5 cells. However, IFN-α can somewhat inhibit HCV replication in the absence of STAT1. It seems that this inhibition is mediated by STAT2 and IRF9, but not STAT3 or STAT6. Meanwhile, IFN-λ inhibits HCV replication only through a STAT1-dependent pathway; while STAT1 inactivation fully suppresses the IFN-λ antiviral activity. According to the results, the induction of ISGs, like PKR and IRF9, by IFN-λ was inhibited by the knockout of STAT1 in HCV-infected cells [84].
Viral evasion
Viruses use immune evasion mechanisms to increase replication and counter host immune surveillance. CRISPR/Cas has been used to study several of such mechanisms. For instance, to investigate the effects of RNase L on ZIKV infection, CRISPR-KO was used to knockout targeted host genes involved in the RNase L pathway. The results demonstrated that ZIKV genome was decreased in the infected wild-type cells compared to RNase L KO cells; while the amount of infectious ZIKV released from the wild-type cells were notably higher than the RNase L KO cells. According to investigations, it seems that ZIKV can escape cleavage by RNAse L due to the formation of replication factories in the membrane of the ER. Therefore, ZIKV genomes resist RNAse L cleavage in such replication factories. While DENV generates replication factories, it is not resistant to RNAse L-mediated cleavage. Consequently, it can be said that this mechanism is specific to ZIKV within the flaviviruses [85]. Studies have identified the interaction of an inactive form of RNase L with actin cytoskeleton to reorganize cellular framework during viral infection [86]. Accordingly, in a recent study, they investigated the role of RNase L during Zika virus infection, and the results indicated the proviral role of inactive RNase L during ZIKV infection. ZIKV induces cytoskeletal remodeling during infection to form replication factories (RFs); the absence of RNase L results in defective remodeling of microtubules. In general, it can be concluded that ZIKV exploits the interaction between RNase L and the cytoskeleton to facilitate ER rearrangement to create RFs, promoting ZIKV production [87].
CRISPR/Cas antiviral strategies against Flaviviridae viruses
CRISPR/Cas technology is a growing field in the prevention and treatment of viral infections. The outbreaks of Flaviviridae members in different parts of the world in recent years emphasize the need for innovative methods of vector control which is a strategy used to limit the transmission rate of these viruses. Furthermore, CRISPR/Cas tools are being used to generate gene drives that can potentially decrease mosquito populations. The use of gene drives for mosquito control has attracted much attention in the recent years, as the ease of production of CRISPR-based gene drive systems sets them apart from other methods [88, 89]. There have been multiple methods introduced in CRISPR-based gene drive systems, which include: suppression drives, through which a weakening gene is inflicted in a target population, limiting their activity or even eradicating the targets; and Modification drives, by which the target population is altered in a desired way (e.g. in the setting of malaria control the mosquitoes are altered not to be capable of transmitting the disease) [90].
CRISPR/Cas9
In general, CRISPR-based antiviral approaches include the inactivation of genes involved in the progression of viral infections and include two modes of knock‐out or knock‐down of viral genes or relevant host factors. Some fundamental challenges in using this system include the following: There is a possibility of off-target mutagenesis, and Cas9/gRNA expression level and duration are also significant factors. Delivering the CRISPR/Cas9 system in vivo efficiently and safely is a considerable clinical challenge [91, 92]. One of the other challenges is the human body’s immunogenicity against the Cas9 protein, which is derived from bacteria [93]. Another important point in CRISPR-Cas9-based therapeutic is related to genome repair or rearrangement processes after double-stranded breaks, which may lead to unexpected mutations [94]. On the other hand, the canonical CRISPR/Cas9 is unable to perform its editing function for the RNA virus genome. The Cpf1 and C2c2/Cas13 among all CRISPR/Cas systems are the ones able to be designed to target the RNA virus genome [95]. The Cas9 endonuclease from Francisella novicida (FnCas9) has also been reported to target endogenously transcribed mRNA and thus regulate gene expression (96). Today, most HCV infections can be treated with appropriate pharmacological interventions. Nevertheless, we may face drug-resistant mutant HCV variants, so the current anti-HCV regimen may not be effective in the future. Therefore, the CRISPR/Cas9-mediated disruption of the HCV genome may be suggested as an anti-HCV strategy. In a study using FnCas9, the HCV RNA genome was targeted in eukaryotic cells, which resulted in the inhibition of viral protein production. In fact, by targeting the 5′-and 3′-UTR of the HCV genomic + ssRNA, virus inhibition was observed due to the blockade of viral RNA translation and viral replication machineries [97].
CRISPR/Cas13
The application of the CRISPR/Cas13a (known previously as C2c2) system to RNA editing has expanded [27, 98]. For example, in a study, Cas13a was reported to target HCV internal ribosomal entry site (IRES), reducing the HCV RNA replication and translation. Thus, using IRES-specific crRNAs, Cas13a can suppress HCV more efficiently in huh-7.5 cells [99]. Moreover, Li et al. [100] hypothesized that the CRISPR/Cas13a system could suppress DENV infection by degrading viral RNA genome or by mutagenizing crucial genomic elements; therefore, they adapted the CRISPR-Cas13a system to DENV and discovered a CRISPR RNA (crRNA) that was able to suppress DENV replication in the cell culture system by targeting the NS3 gene. Another study used a novel strategy using CRISPR/Cas13 against RNA viruses. They indicated that virus-like particles (VLP) could be used to deliver PspCas13b RNP to primary human target cells to suppress dengue virus infection effectively. Shortening the spacer length of crRNA in the range of 18–26 nts improved CRISPR/Cas13b knockdown activity without compromising crRNA processing or multiplex targeting capability [101]. On the other hand, Chen et al. aimed to develop an anti-ZIKV system using CRISPR/Cas13b in mammalian cells. They first generated a cell line susceptible to ZIKV infection and a reporter system, then designed fourteen crRNAs, five of which were effective in targeting conserved regions of the ZIKV genome. Such studies, which try to develop new methods of inhibiting RNA viruses, increase the hopes of using the CRISPR/Cas13 system as a new therapeutic approach in the near future [102]. The CRISPR/Cas13b technology offers tempting advantages for therapeutic purposes. Cas13b can target multiple sites at once, which significantly lowers the chance of viruses esca** the immune system. Instead of exploring the biological characteristics of viruses which are required to produce traditional antiviral drugs, crRNA can be designed just by comprehending the virus genome sequence. CRISPR/Cas13b target-cleavage of RNA is a safer alternative since it is not permanently inherited [103].
Conclusion
Recently, CRISPR/Cas9 has revolutionized the study of host-virus biology. CRISPR/Cas technology is utilized to improve our knowledge of how viruses exploit their hosts, as well as to develop new antiviral therapies. Particularly, further studies are constantly conducted using this approach to improve our understanding of flavivirus life cycles, leading to the discovery of essential factors. However, there are still numerous obstacles that need to be addressed. Viral host factor requirements may differ based on viral strains and cell types. In this regard, studies investigating host-virus interactions using CRISPR must use clinically and epidemiologically important virus isolates and corroborate the results with numerous strains. It is also possible that host factors vary from one cell type to another; for example, ZIKV, which infects specialized cells like neural stem cells. Therefore, CRISPR screens should be conducted in the proper cell contexts to reveal the involved factors comprehensively [104]]. Undoubtedly, future screens will provide insight into how viruses have evolved to exploit and subvert host functions. In addition, the findings may lead to potential targets for antiviral therapy. Finally, we expect that next generation CRISPR approaches alongside other new technologies will help us better understand complex biological processes.
Availability of data and materials
Not applicable.
Abbreviations
- CRISPR: :
-
Clustered regularly interspaced short palindromic repeats
- CRISPRa: :
-
CRISPR activation
- CRISPRi::
-
CRISPR interference
- CRISPR-KO: :
-
CRISPR knockout
- dCas9: :
-
Dead Cas9
- DENV: :
-
Dengue virus
- DPMS: :
-
Dolichol-phosphate mannose synthase
- EMC: :
-
ER membrane complex
- `ER::
-
Endoplasmic reticulum
- ERAD::
-
Endoplasmic-reticulum-associated degradation
- ERGIC: :
-
ER-Golgi intermediate compartment
- FACS: :
-
Fluorescence-activated cell sorting
- FLAD1::
-
Flavin adenine dinucleotide synthetase 1
- HCV: :
-
Hepatitis C virus
- IFN: :
-
Interferon response
- IFN-λ2::
-
Interferon lambda 2
- ISGs: :
-
Interferon-stimulated genes
- JEV::
-
Japanese encephalitis virus
- MAGT::
-
Magnesium transporter
- NGS: :
-
Next-generation sequencing
- NPs: :
-
Neural progenitors
- OAS: :
-
Oligoadenylate synthetases
- OST: :
-
Oligosaccharyltransferase
- RACK1: :
-
Receptor for activated protein C kinase 1
- RFK: :
-
Riboflavin kinase
- RLRs: :
-
RIG-I-like receptors
- RNAi: :
-
RNA interference
- RhoV::
-
Ras homolog family member V
- sgRNA: :
-
Single-guide RNA
- SPC: :
-
Signal peptidase complex
- SPs::
-
Signal peptides
- STAT::
-
Signal transducer and activator of transcription
- TBEV::
-
Tick-borne encephalitis virus
- TMDs: :
-
Transmembrane domains
- TMEM41B: :
-
Transmembrane protein 41B
- TRAP: :
-
Translocon-associated protein
- TRIM26::
-
Tripartite motif containing 26
- UBE2J1: :
-
Ubiquitin-conjugating enzyme E2 J1
- VLP: :
-
Virus-like particles
- VMP1: :
-
Vacuole membrane protein 1
- VPs: :
-
Vesicle packets
- WNV: :
-
West Nile virus
- WWTR1::
-
WW domain containing transcription regulator 1
- YFV: :
-
Yellow fever virus
- ZIKV: :
-
Zika virus
References
Barrows NJ, Campos RK, Liao K-C, Prasanth KR, Soto-Acosta R, Yeh S-C, et al. Biochemistry and molecular biology of flaviviruses. Chem Rev. 2018;118(8):4448–82.
Alazard-Dany N, Denolly S, Boson B, Cosset F-L. Overview of HCV life cycle with a special focus on current and possible future antiviral targets. Viruses. 2019;11(1):30.
Kuno G, Chang G-JJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72(1):73.
Heymann DL, Hodgson A, Freedman DO, Staples JE, Althabe F, Baruah K, et al. Zika virus and microcephaly: Why is this situation a PHEIC? The Lancet. 2016;387(10020):719–21.
Sikka V, Chattu VK, Popli RK, Galwankar SC, Kelkar D, Sawicki SG, et al. The emergence of Zika virus as a global health security threat: a review and a consensus statement of the INDUSEM Joint Working Group (JWG). J Global Infect Dis. 2016;8(1):3.
Saiz J-C, Vázquez-Calvo Á, Blázquez AB, Merino-Ramos T, Escribano-Romero E, Martin-Acebes MA. Zika virus: the latest newcomer. Front Microbiol. 2016;7:496.
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013;496(7446):504–7.
Monath TP, Vasconcelos PF. Yellow fever. J Clin Virol. 2015;64:160–73.
Gardner CL, Ryman KD. Yellow fever: a reemerging threat. Clin Lab Med. 2010;30(1):237–60.
Samaan Z, McDermid Vaz S, Bawor M, Potter TH, Eskandarian S, Loeb M. Neuropsychological impact of West Nile virus infection: an extensive neuropsychiatric assessment of 49 cases in Canada. PLoS ONE. 2016;11(6):e0158364.
DeBiasi RL, Tyler KL. West Nile virus meningoencephalitis. Nat Clin Pract Neurol. 2006;2(5):264–75.
Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Ewing D, Mazowiecki M, et al. West Nile virus—associated flaccid paralysis. Emerg Infect Dis. 2005;11(7):1021.
Collins MH, Metz SW. Progress and works in progress: update on flavivirus vaccine development. Clin Ther. 2017;39(8):1519–36.
Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535(7610):159–63.
Perreira JM, Meraner P, Brass AL. Functional genomic strategies for elucidating human–virus interactions: will CRISPR knockout RNAi and haploid cells? Adv Virus Res. 2016;94:1–51.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7.
Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–4.
Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–4.
Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45(1):273–97.
Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151(3):653–63.
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–7.
Barrangou R. Diversity of CRISPR-Cas immune systems and molecular machines. Genome Biol. 2015;16(1):1–11.
Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468(7320):67–71.
Tang L. Exploring class 1 CRISPR systems. Berlin: Nature Publishing Group; 2019.
Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat Rev Microbiol. 2017;15(3):169–82.
Li Y, Peng N. Endogenous CRISPR-Cas system-based genome editing and antimicrobials: review and prospects. Front Microbiol. 2019;10:2471.
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR–Cas13. Nature. 2017;550(7675):280–4.
Cox DB, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, et al. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–27.
Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60(3):385–97.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173(3):665–76.
Xu C, Zhou Y, **ao Q, He B, Geng G, Wang Z, et al. Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes. Nat Methods. 2021;18(5):499–506.
Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356(6336):438–42.
Yang Y, Xu J, Ge S, Lai L. CRISPR/Cas: advances, limitations, and applications for precision cancer research. Front Med. 2021;8:649896.
Zhang B. CRISPR/Cas gene therapy. J Cell Physiol. 2021;236(4):2459–81.
Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25(2):249–54.
Liu J-Q, Li T. CRISPR-Cas9-mediated loss-of-function screens. Front Life Sci. 2019;12(1):1–13.
Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9(4):363–6.
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA–guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–9.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55.
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer PR, Lin E, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12(4):326–8.
Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159(3):647–61.
Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159(3):635–46.
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–8.
Grove J, Marsh M. The cell biology of receptor-mediated virus entry. J Cell Biol. 2011;195(7):1071–82.
Chu J, Ng M. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J Virol. 2004;78(19):10543–55.
Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338(1):53–60.
Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80(14):6964–72.
Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–88.
Richardson RB, Ohlson MB, Eitson JL, Kumar A, McDougal MB, Boys IN, et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat Microbiol. 2018;3(11):1214–23.
Lin DL, Cherepanova NA, Bozzacco L, MacDonald MR, Gilmore R, Tai AW. Dengue virus hijacks a noncanonical oxidoreductase function of a cellular oligosaccharyltransferase complex. MBio. 2017;8(4):e00939-e1017.
Ngo AM, Shurtleff MJ, Popova KD, Kulsuptrakul J, Weissman JS, Puschnik AS. The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. Elife. 2019;8:e48469.
Labeau A, Simon-Loriere E, Hafirassou M-L, Bonnet-Madin L, Tessier S, Zamborlini A, et al. A genome-wide CRISPR-Cas9 screen identifies the dolichol-phosphate mannose synthase complex as a host dependency factor for dengue virus infection. J Virol. 2020;94(7):e01751-e1819.
Brugier A, Hafirrassou ML, Pourcelot M, Baldaccini M, Kril V, Couture L, et al. RACK1 associates with RNA-binding proteins vigilin and SERBP1 to facilitate dengue virus replication. J Virol. 2022;96(7):e01962-e2021.
Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, et al. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016;16(1):232–46.
Hoffmann H-H, Schneider WM, Rozen-Gagnon K, Miles LA, Schuster F, Razooky B, et al. TMEM41B is a pan-flavivirus host factor. Cell. 2021;184(1):133–48.
Shue B, Chiramel AI, Cerikan B, To T-H, Frölich S, Pederson SM, et al. Genome-wide CRISPR screen identifies RACK1 as a critical host factor for flavivirus replication. J Virol. 2021;95(24):e00596-e621.
Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, et al. A CRISPR-based screen identifies genes essential for West-Nile-virus-induced cell death. Cell Rep. 2015;12(4):673–83.
Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016;535(7610):164–8.
Shirasago Y, Shimizu Y, Tanida I, Suzuki T, Suzuki R, Sugiyama K, et al. Occludin-knockout human hepatic Huh7. 5.1–8-derived cells are completely resistant to hepatitis C virus infection. Biol Pharm Bull. 2016:b15–01023.
Ren Q, Li C, Yuan P, Cai C, Zhang L, Luo GG, et al. A Dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci Rep. 2015;5(1):1–7.
Liang Y, Zhang G, Li Q, Han L, Hu X, Guo Y, et al. TRIM26 is a critical host factor for HCV replication and contributes to host tropism. Sci Adv. 2021;7(2):9732.
Luu AP, Yao Z, Ramachandran S, Azzopardi SA, Miles LA, Schneider WM, et al. A CRISPR Activation screen identifies an atypical rho GTPase that enhances Zika viral entry. Viruses. 2021;13(11):2113.
Wang S, Zhang Q, Tiwari SK, Lichinchi G, Yau EH, Hui H, et al. Integrin αvβ5 internalizes Zika virus during neural stem cells infection and provides a promising target for antiviral therapy. Cell Rep. 2020;30(4):969–83.
Garcia G Jr, Paul S, Beshara S, Ramanujan VK, Ramaiah A, Nielsen-Saines K, et al. Hippo signaling pathway has a critical role in Zika virus replication and in the pathogenesis of neuroinflammation. Am J Pathol. 2020;190(4):844–61.
Shrimal S, Cherepanova NA, Gilmore R, editors. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. In: Seminars in cell & developmental biology; 2015. Elsevier.
Wideman JG. The ubiquitous and ancient ER membrane protein complex (EMC): Tether or not? F1000Research. 2015;4.
Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, et al. Defining human ERAD networks through an integrative map** strategy. Nat Cell Biol. 2012;14(1):93–105.
Moretti F, Bergman P, Dodgson S, Marcellin D, Claerr I, Goodwin JM, et al. TMEM 41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018;19(9):e45889.
Morita K, Hama Y, Mizushima N. TMEM41B functions with VMP1 in autophagosome formation. Autophagy. 2019;15(5):922–3.
Morishita H, Zhao YG, Tamura N, Nishimura T, Kanda Y, Sakamaki Y, et al. A critical role of VMP1 in lipoprotein secretion. Elife. 2019;8:e48834.
Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol. 2004;11(10):957–62.
Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334(6062):1524–9.
Pfeffer S, Dudek J, Schaffer M, Ng BG, Albert S, Plitzko JM, et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat Commun. 2017;8(1):1–9.
Ohlson MB, Eitson JL, Wells AI, Kumar A, Jang S, Ni C, et al. Genome-scale CRISPR screening reveals host factors required for ribosome formation and viral replication. MBio. 2023;14(2):e00127-e223.
Beaulieu-Laroche L, Christin M, Donoghue A, Agosti F, Yousefpour N, Petitjean H, et al. TACAN is an ion channel involved in sensing mechanical pain. Cell. 2020;180(5):956–67.
Malik P, Korfali N, Srsen V, Lazou V, Batrakou DG, Zuleger N, et al. Cell-specific and lamin-dependent targeting of novel transmembrane proteins in the nuclear envelope. Cell Mol Life Sci. 2010;67(8):1353–69.
Li S, Qian N, Jiang C, Zu W, Liang A, Li M, et al. Gain-of-function genetic screening identifies the antiviral function of TMEM120A via STING activation. Nat Commun. 2022;13(1):105.
Turpin J, Frumence E, Harrabi W, Haddad JG, El Kalamouni C, Desprès P, et al. Zika virus subversion of chaperone GRP78/BiP expression in A549 cells during UPR activation. Biochimie. 2020;175:99–105.
Dukhovny A, Lamkiewicz K, Chen Q, Fricke M, Jabrane-Ferrat N, Marz M, et al. A CRISPR activation screen identifies genes that protect against Zika virus infection. J Virol. 2019;93(16):e00211-e219.
Li Y, Muffat J, Javed AO, Keys HR, Lungjangwa T, Bosch I, et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc Natl Acad Sci. 2019;116(19):9527–32.
Hartmann G. Nucleic acid immunity. Adv Immunol. 2017;133:121–69.
Schilling M, Bridgeman A, Gray N, Hertzog J, Hublitz P, Kohl A, et al. RIG-I plays a dominant role in the induction of transcriptional changes in Zika virus-infected cells, which protect from virus-induced cell death. Cells. 2020;9(6):1476.
Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, et al. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci. 2016;113(8):2241–6.
Yamauchi S, Takeuchi K, Chihara K, Honjoh C, Kato Y, Yoshiki H, et al. STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-λ but not by interferon-α. Sci Rep. 2016;6(1):1–11.
Whelan JN, Li Y, Silverman RH, Weiss SR. Zika virus production is resistant to RNase L antiviral activity. J Virol. 2019;93(16):e00313–9.
Malathi K, Siddiqui M, Dayal S, Naji M, Ezelle H, Zeng C, et al. RNase L interacts with Filamin A to regulate actin dynamics and barrier function for viral entry. MBio. 2014;5:e02012.
Whelan JN, Parenti NA, Hatterschide J, Renner DM, Li Y, Reyes HM, et al. Zika virus employs the host antiviral RNase L protein to support replication factory assembly. Proc Natl Acad Sci. 2021;118(22):e2101713118.
Li M, Yang T, Kandul NP, Bui M, Gamez S, Raban R, et al. Development of a confinable gene drive system in the human disease vector Aedes aegypti. Elife. 2020;9:e51701.
Adelman ZN, Tu Z. Control of mosquito-borne infectious diseases: sex and gene drive. Trends Parasitol. 2016;32(3):219–29.
Bier E. Gene drives gaining speed. Nat Rev Genet. 2022;23(1):5–22.
Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat Rev Genet. 2016;17(5):300–12.
Wei T, Cheng Q, Farbiak L, Anderson DG, Langer R, Siegwart DJ. Delivery of tissue-targeted scalpels: opportunities and challenges for in vivo CRISPR/Cas-based genome editing. ACS Nano. 2020;14(8):9243–62.
Mehta A, Merkel OM. Immunogenicity of Cas9 protein. J Pharm Sci. 2020;109(1):62–7.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8.
Chen S, Yu X, Guo D. CRISPR–Cas targeting of host genes as an antiviral strategy. Viruses. 2018;10(1):40.
Sampson TR, Saroj SD, Llewellyn AC, Tzeng Y-L, Weiss DS. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497(7448):254–7.
Price AA, Sampson TR, Ratner HK, Grakoui A, Weiss DS. Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc Natl Acad Sci. 2015;112(19):6164–9.
Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573.
Ashraf MU, Salman HM, Khalid MF, Khan MHF, Anwar S, Afzal S, et al. CRISPR-Cas13a mediated targeting of hepatitis C virus internal-ribosomal entry site (IRES) as an effective antiviral strategy. Biomed Pharmacother. 2021;136:111239.
Li H, Wang S, Dong X, Li Q, Li M, Li J, et al. CRISPR-Cas13a cleavage of dengue virus NS3 gene efficiently inhibits viral replication. Mol Ther-Nucleic Acids. 2020;19:1460–9.
Singsuksawat E, Onnome S, Posiri P, Suphatrakul A, Srisuk N, Nantachokchawapan R, et al. Potent programmable antiviral against dengue virus in primary human cells by Cas13b RNP with short spacer and delivery by VLP. Mol Ther-Methods Clin Dev. 2021;21:729–40.
Chen P, Chen M, Chen Y, **g X, Zhang N, Zhou X, et al. Targeted inhibition of Zika virus infection in human cells by CRISPR-Cas13b. Virus Res. 2022;312:198707.
Freije CA, Myhrvold C, Boehm CK, Lin AE, Welch NL, Carter A, et al. Programmable inhibition and detection of RNA viruses using Cas13. Mol Cell. 2019;76(5):826–37.
Simonin Y, Loustalot F, Desmetz C, Foulongne V, Constant O, Fournier-Wirth C, et al. Zika virus strains potentially display different infectious profiles in human neural cells. EBioMedicine. 2016;12:161–9.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
ZR: built the structure, completed the first draft, and prepared the manuscript. AS: critically revised and edited the manuscript, and provided the final draft. HBB: supervised and revised the manuscript. The submitted version was read and approved by the authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ramezannia, Z., Shamekh, A. & Bannazadeh Baghi, H. CRISPR–Cas system to discover host-virus interactions in Flaviviridae. Virol J 20, 247 (2023). https://doi.org/10.1186/s12985-023-02216-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-02216-7