
Abstract
Nerve injury-induced chronic pain has been an urgent problem for both public health and clinical practice. While transition to chronic pain is not an inevitable consequence of nerve injuries, the susceptibility/resilience factors and mechanisms for chronic neuropathic pain after nerve injuries still remain unknown. Current preclinical and clinical studies, with certain notable limitations, have shown that major histocompatibility complex class II–restricted T helper (Th) cells is an important trigger for nerve injury-induced chronic tactile allodynia, one of the most prevalent and intractable clinical symptoms of neuropathic pain. Moreover, the precise pathogenic neuroimmune interfaces for Th cells remain controversial, not to mention the detailed pathogenic mechanisms. In this review, depending on the biology of Th cells in a neuroimmunological perspective, we summarize what is currently known about Th cells as a trigger for chronic tactile allodynia after nerve injuries, with a focus on identifying what inconsistencies are evident. Then, we discuss how an interdisciplinary perspective would improve the understanding of Th cells as a trigger for chronic tactile allodynia after nerve injuries. Finally, we hope that the expected new findings in the near future would translate into new therapeutic strategies via targeting Th cells in the context of precision medicine to either prevent or reverse chronic neuropathic tactile allodynia.
Similar content being viewed by others

Background
Neuropathic pain is a debilitating category of pathological pain caused by a heterogeneous repertoire of lesions or diseases of the somatosensory system, which can result in either heightened or disordered transmission of sensory signals into the spinal cord and the brain [1]. The most common conditions associated with neuropathic pain involve injuries to peripheral nervous system (PNS), such as mechanical nerve trauma or compression (painful radiculopathies included), postherpetic neuralgia (PHN), painful diabetic neuropathy (PDN), chemotherapy-induced peripheral neuropathy (CIPN), and trigeminal neuralgia (TGN) [1]. This chronic disease typically manifests as an increased or altered sensitivity to mechanical or thermal stimuli (hyperalgesia or allodynia). It is resistant to conservative medical management and significantly decreases the quality of life [2]. Therefore, the effective therapeutic options to either prevent or reverse chronic neuropathic pain is a critical unmet need for both public health and clinical practice [1].
It is well-recognized that, even with the same nerve injuries, some individuals go on to develop chronic neuropathic pain, while others experience a resolution of acute neuropathic pain [3, 4]. However, the susceptibility/resilience factors and mechanisms for chronic neuropathic pain after nerve injuries are just at the beginning to be elucidated [4, 5]. This is a crucial scientific problem because elucidating why and how individuals develop or withstand chronic neuropathic pain after nerve injuries will pave the way of translational pain medicine for the development of new therapeutic strategies in the context of precision medicine to either prevent or reverse chronic neuropathic pain [5].
Tactile allodynia is one of the most prevalent and intractable clinical symptoms of neuropathic pain after nerve injuries [2, 6]. Mechanistically, tactile allodynia ultimately arises because of the disordered transmission of touch sensory signals, mainly from myelinated low-threshold mechanoreceptors (LTMRs), into the nociceptive circuitry at varying levels of the nervous system, resulting in the erroneous perception of touch as pain [6,7,8]. Neuronal plasticity has been shown as the fundamental process for the initiation, chronification, and maintenance of neuropathic pain [9]. Interestingly, pain research in the past two decades has established that neuroinflammation is a key driving force for neuronal plasticity underlying neuropathic pain, including tactile allodynia [10, 11]. While many of current studies focus on the onset of neuropathic pain during the acute phase following nerve injuries [5], accumulating evidence indicates that distinct mechanisms engage when neuropathic pain progresses [10]. For instance, after nerve injuries, while microglia are rapidly activated to promote the onset of neuropathic pain [12], astrocytes are activated with a delay of several days or weeks and drive the chronification and maintenance of neuropathic pain rather than its initiation [13]. In this review, we therefore give a brief overview of the key findings regarding the neurobiological and immunological mechanisms for how tactile allodynia gets stuck after nerve injuries in susceptible individuals.
Major histocompatibility complex class II (MHCII)-restricted T helper (Th) cells, a pivotal category of the heterogeneous T cell population [14], have been shown as an important trigger for chronic tactile allodynia after nerve injuries [15]. However, certain limitations for the current state of preclinical and clinical evidences must be faced up to. Moreover, it remains controversial as to where along the neuroaxis Th cells act as a trigger for chronic tactile allodynia after nerve injuries [15]. The uncertainty of the pathogenic neuroimmune interfaces for Th cells presents an inescapable obstacle for further insights into the detailed mechanisms for Th cells as a trigger for chronic tactile allodynia after nerve injuries. The essential reason underlying the current dispirited state is that these neuroimmune studies are, more often than not, designed by and for neuroscientists themselves. The inadequate adoption of immunological perspective, nomenclature, and techniques of Th cells makes the current evidences inconsistent.
With the simple import of immunological nomenclature and techniques, recent studies have rapidly provided strong contrasting evidence to the prevailing notions and have demonstrated the absence of blood-derived monocyte infiltration into the spinal cord dorsal horns (SC-DHs) after nerve injuries [16,17,18,19,20]. Therefore, we focus here on an interdisciplinary perspective, Th cell neuroimmunology in particular, that would benefit neuroscientists who desire deeper insights into pain neuroimmunology. First, we introduce the biology of Th cells in a neuroimmunological perspective. Then, we summarize what is currently known about Th cells in the development of chronic tactile allodynia after nerve injuries, with a focus on identifying what inconsistencies are evident. Finally, we discuss how an interdisciplinary perspective would result in a more comprehensive understanding of the detailed roles and mechanisms of Th cells as a trigger for chronic tactile allodynia after nerve injuries. This knowledge would ultimately herald a new era for either preventing or reversing chronic neuropathic tactile allodynia via targeting Th cells in the context of precision medicine.
The acute to chronic transition of tactile allodynia after nerve injuries
The sense of touch (pressure is not considered in this review) is essential for daily activities throughout our lives, as it provides the vital real-time information about the nature of our physical environment [21]. In the periphery, touch sensations are mainly conveyed by myelinated LTMRs, which are upstream-activated by their innervated mechanosensory end organs. Then, the innocuous touch input from the periphery is transmitted to deep dorsal horn (DH) circuits within the spinal cord (SC) and ultimately relayed to the somatosensory cortex for the perception of touch [21]. At the same time, myelinated LTMR inputs are feedforward gated to prevent the activation of spinal pain transmission neurons (PTNs) within the superficial DH via physiologically silent, dorsally directed neural microcircuits (Fig. 1) [6, 22]. Therefore, touch is not perceived as pain under physiological conditions.

The SC-DH circuit for the transformation of touch into pain (i.e., tactile allodynia) after nerve injuries. a Neural circuit framework of neuropathic tactile allodynia based on the gate control theory of pain. b Schematic illustration of the SC-DH circuit for tactile allodynia after nerve injuries. While static tactile allodynia evoked by von Frey hairs is dependent on VT3− excitatory interneurons, morphine-resistant, dynamic tactile allodynia evoked by brushing is dependent on VT3+ excitatory interneurons. Moreover, while TAC1−NK1R+ PTNs are sufficient for reflexive defensive reactions, TAC1+NK1R+ PTNs are necessary for sustained pain-associated co** behaviors. CCK, cholecystokinin; CR, calretinin; CST, corticospinal tract; DRG, dorsal root ganglion; EN, excitatory interneuron; IN, inhibitory interneuron; LTMR, low-threshold mechanoreceptor; MRGPRD, MAS related GPR family member D; MSC, myelinated Schwann cell; NK1R, neurokinin 1 receptor; PKCγ, protein kinase C gamma; PSDCN, postsynaptic dorsal column neuron; PTN, pain transmission neuron; RORα, retinoid receptor-related orphan receptor; SC-DH, spinal cord dorsal horn; SCTN, spino-cervical tract neuron; SOM, somatostatin; TAC1, preprotachykinin 1; TRPV1, transient receptor potential cation channel, subfamily V, member 1; VT3, vesicular glutamate transporter 3
Following nerve injuries, the innocuous touch become painful. The erroneous perception of touch as pain involves robust neuronal changes in the key SC-DH circuits responsible for tactile allodynia in the form of “modulation” or “modification” (Fig. 2a) [6, 9]. Aberrant primary afferent input and altered descending supraspinal input collectively drive neuronal plasticity in the SC-DH [23, 24]. The resulting central sensitization (including facilitation and disinhibition) within the SC-DH circuits opens or overcomes the inhibitory gates for LTMR inputs, thus allowing innocuous touch stimuli to directly activate physiologically silent and polysynaptic neural microcircuits linked to pain sensation (Fig. 1) [22,23,24]. Moreover, during this painful transformation of touch, neuroinflammation across the somatosensory pathway has been shown to play an active role [6, 10]. Detailed information about the current state of evidence for the transformation of touch into pain after nerve injuries has been summarized in recent review articles [6, 25].

The acute to chronic transition of tactile allodynia after nerve injuries. a Schematic illustration of the continuum of neuronal plasticity ranging from “activation” to “modulation” and “modification” for the development of distinct pain states. Detailed forms of neuronal plasticity for PSNs and SC-DH neurons are shown in brief. b Schematic summary of the typical “modification” processes in the SC-DH circuit underlying the development of chronic tactile allodynia after nerve injuries. CCK2, cholecystokinin type 2 receptor; EN, excitatory interneuron; F, function; IL-6, interleukin-6; IN, inhibitory interneuron; MCP-3, monocyte chemotactic protein-3; MOR, μ opioid receptor; PKCγ, protein kinase C gamma; PSN, primary sensory neuron; PTM, post-translational modification; PV, parvalbumin; RVM, rostral ventromedial medulla; S, structure; SC-DH, spinal cord dorsal horn
However, how tactile allodynia becomes chronic after nerve injuries is one of the most important but still unclear questions at this moment. Therefore, we instead give particular attention to the key findings regarding the neurobiological and immunological mechanisms for the acute to chronic transition of tactile allodynia in susceptible individuals after nerve injuries (Fig. 2b). In essence, transition to chronic tactile allodynia after nerve injuries is the “modification” process establishing stable structural/functional microcircuits for persistent but disordered transmission of touch sensory signals into the nociceptive circuitry in the SC-DH (Fig. 2a) [3, 9].
Local modifications within the SC-DH have been shown to underlie the development of this chronic condition. The most prominent example is persistent local disinhibition, in the form of the robust pruning of inhibitory synapses from parvalbumin (PV) interneurons onto PKCγ excitatory interneurons [26] or the excitotoxic cell death of γ-aminobutyric acid (GABAergic) inhibitory interneurons [27]. Modifications in descending supraspinal inputs are also engaged. On the one hand, nerve injuries can result in significant loss of descending pontospinal noradrenergic inhibitory fibers within the SC-DH [28]. On the other hand, nerve injuries lead to delayed enhancement of descending facilitation from “On” cells in the rostral ventromedial medulla (RVM), which co-express mu opioid receptor (MOR) and cholecystokinin type 2 receptor (CCK2) [37].

The definition and identification for Th cells from the perspective of thymic development of Th cells. a Schematic summary of the development of Th cells in the thymus cortex and medulla. b Schematic illustration of Th cells versus innate T cells among CD4+ αβ T cells. Th cells are, in the strict definition, MHCII-restricted CD4+ αβ T cells and PLZF negative. The identification methods for Th cells are shown. DC, dendritic cell; PLZF, promyelocytic leukemia zinc finger; TEC, thymic epithelial cell; TRA, tissue-restricted antigen; UEA, ubiquitous-expressed antigen
Hence, Th cells are, in the strict definition, MHCII-restricted CD4+ αβ T cells (Fig. 3b). Since there are few, if any, CD4+ γδ T cells [38], Th cells are conventionally named as CD4+ T cells and identified as CD3+ CD4+ cells by histology in situ or flow cytometry in single-cell suspension (Fig. 3b). However, CD4+ αβ T cells or CD4+ T cells are composed of not only a major population of MHCII-restricted Th cells, but also a minor population of innate CD4+ αβ T cells, including CD1d-restricted natural killer T cells (NKTs) (Fig. 3b) [39]. There are currently no unique molecular markers to directly distinguish Th cells from innate CD4+ αβ T cells among CD4+ αβ T cells or CD4+ T cells. The main reason for this technical challenge is that Th cells and innate CD4+ αβ T cells often express a common or similar set of transcriptional factors, cytokines, and surface molecules [40]. Therefore, the most accurate identification of Th cells requires combinatorial use of CD4/αβ TCR or CD4/CD3 double labeling as Th cell detection method and MHCII knockout animals as Th cell depletion method (Fig. 3b). However, this identification strategy is achievable only in preclinical studies, especially in mice [41], rather than clinical studies. In the case that the cognate antigens are known, MHCII tetramer staining is the ideal methods to identify Th cells in clinical studies (Fig. 3b) [42].
The promyelocytic leukemia zinc finger (PLZF) is a specific key transcription factor for the development of innate CD4+ αβ T cells but not for Th cells [43,44,45,46]. It has been reported that PLZF expression is highly specific to innate T cells, rather than conventional T cells (including Th cells). Furthermore, PLZF expression cannot be induced de novo in conventional T cells via TCR-mediated activation or inflammation [47]. Therefore, PLZF could be used as a potential effective molecular biomarker to directly distinguish PLZF− Th cells from PLZF+ innate CD4+ αβ T cells for technical practice (Fig. 3b). However, this potential molecular biomarker requires further validation of the ubiquitous expression of PLZF in all the innate CD4+ αβ T cells.
How do Th cells function?
Following maturation in the thymus, naïve Th cells enter into the blood circulation and traffic between secondary lymphoid organs (the spleen and lymph nodes) and the blood. Upon antigen stimulation, naïve Th cells are confronted with three integrated tasks to fulfill their critical roles in immunity (Fig. 4a). First, naïve Th cells must decide whether to turn on or off an immune response. Second, if an immune response is turned on, the proper effector classes of Th cells must be decided before functioning via the manner of contact/secretion in the inflamed target tissues. Third, memory Th cells, either effector or central, should be formed to remember past encounters.

The biology of Th cells at the periphery. a Schematic illustration of the activation of Th cells upon antigen stimulation at the periphery. According to “the second touch” hypothesis, full Th polarization needs a second antigen presentation to primed Th cells by APCs in the inflamed tissues. b Schematic summary of peripheral tolerance mechanisms against autoreactive Th cells, which escape from thymic negative selection. Ag, antigen; AICD, activation-induced cell death; APC, antigen-presenting cell; DC, dendritic cell; pTh, primed Th cell
In secondary lymphoid organs, through dynamic contact with DCs in the form of immunological synapses, naïve Th cells are activated and expanded by two signals from the same DC: one from αβ TCR/CD4 engagement with antigenic peptide in the context of MHCII and the other one through CD28 engagement by CD80 and/or CD86 (Fig. 4a) [34, 48]. Activated Th cells are checked for survival and then acquire an array of activation markers [34].
Upon activation, the proper effector classes of Th cells must be decided to fulfill their essential role in immune responses (Fig. 4a). The main subsets of Th cells are Th1, Th2, Th17, inducible Treg (iTreg), follicular T helper (TFH), and follicular Treg (fTreg) cells [14, 34]. Traditional view holds that activated Th cells are fully polarized to particular Th subsets directly in secondary lymphoid organs. The cytokine milieu that is generated by APCs is an important factor that influences the differentiation of activated Th cells [14, 34, 48]. However, antigen-experienced Th cells acquire peripheral homing receptors soon after their activation [49]. This forces these primed Th cells to rapidly return to the tissue where the antigen resides, which would result in incomplete polarization of activated Th cells in secondary lymphoid organs per se [50]. Accumulating evidence indicated that additional antigen presentation to primed Th cells takes place in the non-lymphoid tissues where the antigen resides [51, 52]. This led to the proposal of “the second touch hypothesis,” which argued that full Th cell polarization into the proper effector classes requires a second antigen presentation to primed Th cells by APCs, either DCs or macrophages, in the inflamed tissues [34].
Autoreactive Th cells, which escape from thymic negative selection, are further checked robustly at the periphery to prevent the development of autoimmunity (Fig. 4b) [37]. First, autoreactive Th cells can ignore self-antigens that are not adequately presented by DCs. The inadequate presentation may result from the low accessible amount of self-antigens per se or the biased peptide presentation of a particular MHCII haplotype. Then, if self-antigens are adequately presented by DCs, autoreactive Th cells can be checked via tuning the fates of activation. Although mature DCs sample and present self-antigens to autoreactive Th cells, these Th cells may either depleted via activation-induced cell death (AICD) or inactivated to become unresponsive or anergic Th cells. Immature or tolerogenic DCs also continuously sample and present self-antigens to autoreactive Th cells, but bias Th cell differentiation into iTreg cells to suppress autoimmunity. Last but not the least, after their activation by mature DCs, autoreactive Th cells can be checked at the point of polarization to avoid autoimmunity. Low or intermediate TCR signal intensity relative to the threshold of Th cell activation has been reported as a determinant for iTreg/Th2 cell polarization to suppress autoimmunity [53, 54].
How to methodologically define Th cell effector subsets?
At present, phenotypic characterization of Th cell effector subsets is defined by the signature cytokines or chemokine receptors that the Th cell subset secretes or expresses and by the master transcription factors upon which the Th cell subset arises [14, 34]. In detail, among Th cells, Th1 cells are identified as T-bet+IFNγ+ cells, Th2 cells as Gata3+IL-4+ cells, Th17 cells as RORγ+IL-17+ cells, iTreg cells as Foxp3+TGFβ+ cells, TFH cells as Bcl-6+CXCR5+ cells, and fTreg cells as Foxp3+CXCR5+ cells (Fig. 4a). Over the years, flow cytometry for immune cell suspensions and histological assays in situ with selected molecular markers have established themselves for phenotypic characterization of Th cell subsets [55].
However, additional complexity in the differentiation of Th cells is becoming evident. On the one hand, it remains controversial whether each subset of Th cells is firmly fixed or remains plastic [14]. For instance, under some circumstances, Th17 cells can also produce IFNγ, the signature cytokine of Th1 cells [14, 56]. More importantly, the effector functions of Th17 cells in autoimmune diseases require their transdifferentiation into Th1-like cells via metabolic reprogramming [56]. The co-expression of Foxp3 and RORγt, the master transcription factor for iTreg and Th17 cells, respectively, has been seen in the same Th cell. Consequently, it seems that some iTreg cells can be induced to transdifferentiate into Th17 cells [14]. On the other hand, accumulating evidence suggests diverse functional states of a particular subset of Th cells. For instance, in vitro polarized Th17 cells can either cause severe autoimmune responses upon adoptive transfer, or have little or no effect in inducing autoimmune disease [57]. Therefore, the complexity of Th cell polarization renders doubtful the use of the selected but limited molecular markers to define a particular subset of Th cells [55].
How do Th cells enter the CNS?
As stated in the “Preclinical evidences” section, the effector immune functions of Th cells, excepting TFH and fTreg cells, require the homing of primed Th cells back to the inflamed target tissues where the corresponding antigen resides. During immune responses in the central nervous system (CNS), traditional views hold that Th cells infiltrate the inflamed nervous tissues via the dysfunctional blood-brain barrier (BBB). However, a growing body of evidence indicates that the infiltration of pathogenic Th cells into the CNS parenchyma is likely secondary to their infiltration into the cerebrospinal meninges during neuroinflammation [58]. Over the past decades, accumulating anatomical and histological studies of the cerebrospinal meninges provide the structural basis for the cerebrospinal meninges as a critical neuroimmune interface for both beneficial and detrimental cross-talks between adaptive immunity and the CNS during homeostasis and diseases (Fig. 5a) [58,59,60].

The cerebrospinal meninges in the pathogenesis of experimental autoimmune encephalomyelitis, a Th cell-mediated autoimmune disease. a Schematic illustration of the anatomy and histology of cerebrospinal meninges from the neuroimmunological point of view. The communication of the glymphatic system and the cerebrospinal meninges is shown. b The infiltration of pathogenic Th cells into the cerebrospinal meninges before their infiltration into the CNS parenchyma. Ag, antigen; APC, antigen-presenting cell; CSF, cerebrospinal fluid; DC, dendritic cell; FRC, fibroblastic reticular cell; mamo, macromolecules; mo, monocyte; mø, macrophage; MVEC, meningeal vascular endothelial cell; SAS, subarachnoid space; ThE, effector Th cell; TLT, tertiary lymphoid tissue
For instance, in rat experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, the spinal leptomeninges are shown as a checkpoint where activated Th cells are licensed to enter the CNS parenchyma. Here, circulating effector Th cells have been shown to get arrested to leptomeningeal vessels and immediately monitor the luminal surface, crawling preferentially against the blood flow (Fig. 5b) [61]. After diapedesis, effector Th cells are anchored in the 3D leptomeningeal network of collagen fibers for effective licensing via leptomeningeal macrophages which effectively present cognate antigens derived from myelin proteins. This meningeal licensing is achieved via antigen-specific physical contacts with resident macrophages, chemokine signaling via CCR5/CXCR3, and integrin signaling (Fig. 5b) [52]. The non-licensed Th cells are preferentially released into the cerebrospinal fluid (CSF), from where they can actively search for damaged nervous tissue areas of cognate antigen availability. Upon arriving at the leptomeninges around these tissue areas, Th cells are licensed there via the same mechanisms mentioned above (Fig. 5b) [52]. Moreover, with the co-operation of meningeal stromal cells, such as fibroblastic reticular cells (FRCs), effector Th cells induce the formation of tertiary lymphoid tissues (TLTs) in the cerebrospinal meninges. The de novo TLTs enable further effector Th cells to reside within the meninges (Fig. 5b) [60].
Emerging roles of Th cells in the transition to chronic tactile allodynia after nerve injuries
In this section, we summarize both clinical and preclinical evidences showing Th cells, the orchestrator of an immune response, as an emerging trigger for chronic tactile allodynia after nerve injuries. Notably, we focus here on primary nerve injuries per se, rather than nerve injuries secondary to autoimmune attacks or infections, such as Guillain-Barre syndrome (GBS) and PHN. In these primarily immune-drived nerve injuries, it is technically difficult to ascertain specific roles of Th cells in the pathogenesis of primary nerve damage versus secondary neuropathic pain.
Clinical evidences
MHCII (Fig. 6a, b) is specifically required for thymic development and peripheral activation of Th cells (Figs. 3a and 4a). Furthermore, MHCII gene polymorphism has been implicated in establishing or breaking central and peripheral tolerance for Th cells via fine tuning the affinity of pMHCII: TCR interactions and the TCR signal intensity (Fig. 6c, d, e) [62,63,64,65,66,67,68]. Therefore, MHCII gene polymorphism is associated with the susceptibility or resistance to Th cell-mediated autoimmunity. In humans, major MHCII isoforms for antigen presentation to Th cells by APCs are from the families of human leukocyte antigen (HLA)-DP, DQ, and DR (Fig. 6b). Recent clinical studies have shown an association of human MHCII gene polymorphism with the susceptibility to chronic neuropathic pain after nerve injuries. In particular, the DQB1*03:02 HLA haplotypes has been shown to have an increased risk for the development of chronic neuropathic pain after inguinal hernia surgery and lumbar disc herniation (LDH) [69]. Likewise, human whole blood transcriptomic profiles showed that the significant upregulation of MHCII antigen presentation pathway genes is associated with the vulnerability of the transition from acute to chronic low back pain, a prevalent form of neuropathic pain [70]. Therefore, these clinical evidences indicate that Th cells may play an initiating role in the development of chronic neuropathic pain, including chronic tactile allodynia.

MHCII gene polymorphism and Th cell-mediated autoimmunity. a, b Schematic illustration of molecular structure of MHCII (a) and genetic structure of MHCII (b). c On particular genomic backgrounds and epigenomic states, MHCII gene polymorphism fine tunes the affinity of pMHCII: TCR interactions and the TCR signal intensity. d, e The role of MHCII gene polymorphism in the susceptibility or resistance of Th cell-mediated autoimmunity through modulating thymus selection and Th polarization. APC, antigen-presenting cell; HLA, human leukocyte antigen; MHC, major histocompatibility complex; pTh, pathogenic Th cells for autoimmunity; TCR, T cell receptor
In support of this suggestion, a growing body of clinical evidences has shown the changes of Th cell numbers and subset patterns in peripheral blood specifically in patients with chronic neuropathic pain after nerve injuries. First, compared with health controls, the counts of CD4+ T cells (presumably Th cells) in patients with LDH were shown to be increased and positively correlated with neuropathic pain intensities [71]. In particular, the numbers of Th17 cells were further shown to be higher in patients with ruptured than non-ruptured lumbar discs and also correlated with neuropathic pain intensities [72]. Second, both central and effector memory CD4+ T cells were shown to be significantly increased in chronic neuropathic pain patients with carpal tunnel syndrome (CTS) and complex regional pain syndrome (CRPS), as compared to healthy controls. The upregulation of memory CD4+ T cells (presumably Th cells) would be an indicator of the history of Th cell activation against a particular antigenic stimulus in CTS patients [73, 74]. To our surprise, however, significant decrease of inflammatory Th1 or Th17 cells and increase of anti-inflammatory Th2 or Treg cells were found in patients with chronic neuropathic pain as compared to healthy controls [75,76,77].
Preclinical evidences
In consistent with clinical studies, preclinical studies further support Th cells as an emerging trigger for chronic tactile allodynia after nerve injuries. As in clinical studies, the MHC gene polymorphism has been seen to influence the susceptibility to this chronic condition after nerve injuries in rats, although whether the major MHC haplotype-specific effect on disease susceptibility is mapped to the MHCII gene region need to be determined in detail [78]. Moreover, during the sub-acute phase after nerve injuries in mice or rats, CD4+ T cells (presumably Th cells), rather than other immune cells (such as CD8+ T cells, B cells, natural killer (NK) cells, and macrophages), were found to be selectively activated by DCs and polarized to IFNγ-positive, inflammatory Th1 cells in peripheral lymphoid organs, such as the spleen and local draining lymph nodes [15, 79,80,81]. The distinct involvement of Th subsets after traumatic nerve injuries in clinical and preclinical studies may due to the plasticity of Th cells, especially Th17 cells (Fig. 4a) [56].
In addition, both CD45+CD4+ lymphocytes (possibly Th cells) and CD45+CD8+ lymphocytes were found to be significantly increased in the blood 7 but not 13 days after high-dose paclitaxel induction in adult male C57BL/6 J mice [82]. However, there were no significant changes for CD3-positive T cells in the blood 7 days after low-dose paclitaxel induction in adult male C57BL/6 J mice [83]. Moreover, for peripheral lymphoid organs, such as the spleen and lymph nodes, CD45+CD4+ lymphocytes and their Th1/Th17/Th2/Treg subsets were not found to be significant changed 7 or 13 days after high-dose paclitaxel and oxaliplatin induction in adult male C57BL/6 J mice, except for transient increase in splenic Th2-like cells and sustained increase in nodal Treg cells for oxaliplatin-treated mice [82]. The discrepancy may arise from the detection timepoints relative to CIPN models and sample locations. These indirect preclinical evidences suggest that Th cells may contribute to the transition to chronic tactile allodynia after nerve injuries.
Accumulating studies targeting Th cells have provided more direct preclinical evidences showing Th cells as an emerging trigger for chronic tactile allodynia after nerve injuries (Table 1). First, studies using genetically immunodeficient mice (including RAG1/2 knockout mice, SCID mice, nude mice or rats, and B cell-deficient mice) and T cell reconstitution have demonstrated that T cells, but not B cells, are essential for the development of chronic tactile allodynia after nerve injuries [79, 80, 84,85,86,87]. Furthermore, passive transfer of nerve injury-conditioned, type 1 polarized T cells (presumably Th1 cells) from heterozygous rats into athymic nude rats restored the level of tactile allodynia in the recipients to that of heterozygous donor rats [80]. Therefore, Th cells, and even Th1 cells, may contribute to the development of chronic tactile allodynia after nerve injuries. Second, the transition from acute to chronic tactile allodynia after nerve injuries was shown to be impaired in CD4 knockout mice, which are specifically devoid of CD4+ cells, but restored in CD4 knockout mice reconstituted with CD4+ cells [79]. This implies that Th cells may contribute to the development of chronic tactile allodynia after nerve injuries, since the majority of CD4+ cells (~ 75%) are CD4+ T cells (presumably Th cells) [79]. It is worth noting that CD8+ but not CD4+ T cell reconstitution in RAG1 knockout mice restored the resolution of acute tactile allodynia which was seen in wild-type mice after paclitaxel or cisplatin induction in adult C57BL/6 male and female mice [83, 88]. Similarly, pharmacological or genetic inhibition of cathepsin S, for which the most important biological function lies in the MHCII antigen presentation pathway and the development and activation of Th cells, significantly attenuated the transition to chronic tactile allodynia after nerve injuries [81]. Furthermore, bolus-adoptive transfer of nerve injury-conditioned splenocytes or splenic CD4+ T cells from wild-type mice into cathepsin S knockout mice or splenectomized mice temporarily restored the level of tactile allodynia in the recipients to that of the donors [81]. Therefore, Th cells were suggested to contribute to the development of chronic tactile allodynia after nerve injuries. Last but the most important, the most direct preclinical evidence has been provided by MHCII knockout mice (specifically devoid of MHCII-restricted Th cells) and show the impaired transition of acute tactile allodynia to a chronic state after nerve injuries [89].
Limitations to clinical and preclinical evidences
Both clinical and preclinical evidences clearly showed that Th cells are an emerging trigger for chronic tactile allodynia after nerve injuries. However, there are several notable limitations to the current state of evidences. We list the most prominent limitations in the following text.
First, the current clinical studies are not rationally designed. They are lack of independent cohorts for prospective studies to validate the results from the retrospective discovery cohorts [69]. For analyzing Th cell events during the sub-acute phase after nerve injuries, the appropriate biomarkers at the corresponding timepoints might have not be carefully selected in these clinical studies. These limitations make the interpretation of the results from these clinical studies very difficult. For example, it remains to be clarified whether the paradoxical Th1/Th17/Treg imbalance seen in patients with chronic neuropathic pain [75,76,77] represents an underlying pathophysiological mechanism or just an epiphenomenon as a result of chronic pain-associated, chronic stress [90].
Second, in preclinical studies, accurate targeting and identification of Th cells is not always achieved. Up to now, only one preclinical study used MHCII knockout mice to specifically deplete Th cells to determine their role in the pathogenesis of tactile allodynia after nerve injuries [89]. Moreover, the assessment of tactile allodynia in current preclinical studies solely relies on the paw withdrawal response in the von Frey hair (VFH) test, which has been recognized as a surrogate of static tactile allodynia. However, dynamic tactile allodynia evoked by brushing stimuli is the more clinically relevant form of tactile allodynia, and the role of Th cells in the development of chronic dynamic tactile allodynia has not been determined so far [23]. Moreover, beyond behavioral tests using the paw withdrawal response, additional tests, such as conditioned place aversion (CPA), have been recognized as a necessity for the full assessment of the complex experience of tactile allodynia [91].
Third, there are some common limitations to both preclinical and clinical studies. T cells have been shown to be involved in the development of tactile allodynia, rather than cold allodynia after nerve injuries in male mice [84]. Therefore, future studies are needed to determine the sensory modality specificity for Th cells as a trigger for chronic tactile allodynia after nerve injuries. More importantly, microglia and Th cells have been suggested to be differently engaged in the development of tactile allodynia after nerve injuries in male versus female mice [92, 93]. However, multiple independent studies imply the involvement of Th cells in the transition to chronic tactile allodynia after nerve injuries in male animals (Table 1). Therefore, it remains in both preclinical and clinical studies to further characterize the complex sexual dimorphism for the role of Th cells in the transition to chronic tactile allodynia after nerve injuries. Another limitations that should be overcome is to ascertain whether the role of Th cells in the transition to chronic tactile allodynia after nerve injuries is independent of the skin phenotypes (glabrous versus hairy) and the properties of nerve injuries, such as the type of involved nerves (spinal versus cranial) and damages (mechanical versus non-mechanical).
The pathogenic neuroimmune interfaces for Th cells as a trigger for chronic tactile allodynia after nerve injuries
In this section, depending on the perspective of the neuroimmunology of Th cells, especially the nomenclatures and techniques, we summarized what is currently known about the pathogenic neuroimmune interfaces for Th cells in the development of chronic tactile allodynia after nerve injuries, with a focus on identifying what inconsistencies are evident (Fig. 7a).

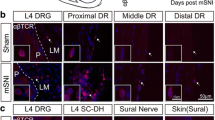
The dorsal root leptomeninges (DRLMs) as the potential neuroimmune interface for Th cells as a trigger for chronic tactile allodynia after nerve injuries. a Schematic summary of current evidences for the infiltration of CD4+ αβ T cells (most possibly Th cells) along the neuroaxis and functional implications of potential Th cell infiltration in the chronification of tactile allodynia after nerve injuries. b Schematic illustration of the anatomy and histology of DRLMs from the neuroimmunological point of view. Region-specific targeting methods for DRLM CD4+ αβ T cells (most possibly Th cells) are shown: [1] LLNe: prior lymphadenectomy to lumbar lymph nodes (LLNs), where CD4+ αβ T cells in DRLMs are derived [2]; chronic intrathecal injection of the suppressive anti-αβTCR antibodies to specifically deplete CD4+ αβ T cells that selectively infiltrate into DRLMs along the neuroaxis after nerve injuries. c The selective infiltration of CD4+ αβ T cells (most possibly Th cells) into lumbar DRLMs along the neuroaxis after adult rat tibial nerve transection (TNT). Here, only the data about DRLMs around the DRG and proximal DR 7 days after nerve injuries is shown for the sake of clarity. d Chronic intrathecal application of the suppressive anti-αβTCR antibodies, which specifically deplete CD4+ αβ T cells (most possibly Th cells) in DRLMs, reduces the development of chronic tactile allodynia after adult rat TNT
Classical pathogenic neuroimmune interfaces
The gray matter of SC-DHs have been classically viewed as an important neuroimmune interface for T cells, including αβ T cells, to initiate the transition to chronic tactile allodynia after nerve injuries [79, 81, 85, 92,93,94,95,96,97]. However, a growing body of evidence doubts about the presence of αβ T cells, or even T cells there. First, a recent study reported little, if any, CD2-positive cells (presumably T cells) in the SC-DHs over 6 weeks after spared nerve injury (SNI) in adult rats [98], with the same experimental settings as in the seminal study concerning the roles of T cell infiltration into the SC-DHs in nerve injury-induced chronic tactile allodynia [85]. Moreover, another recent study reported little, if any, CD3-positive T cells in the SC-DHs 8 and 28 days after SNI in adult C57BL/6 male and female mice [99]. In our previous study, we also did not find any convincing evidences for the presence of αβ T cells in the SC-DHs 1 weeks after tibial nerve transection (TNT) in adult male Sprague-Dawley (SD) rats [55]. Single-cell transcriptomics has been increasingly used to phenotype the subsets or states of immune cells, including Th cells [56, 57]. The molecular landscape of these pathogenic Th cells at single-cell resolution will aid in clarifying how they infiltrate into DRLMs and fulfill their pathogenic role. Another important question to determine is whether the pathogenic role of Th cells here is dependent on MHCII, i.e., antigen specific [130]. In fact, several studies have suggested antigen specificity for these pathogenic Th cells [81, 95, 97, 103]. If this proves to be true in the future, the successful map** of the cognate antigens will aid in deeper insights into the pathogenic mechanisms of Th cells and therefore translate into the development of antigen-specific immunoprevention of chronic tactile allodynia after nerve injuries.
Last, it remains to uncover why Th cells can act as a trigger for chronic tactile allodynia after nerve injuries in susceptible individuals. MHCII gene polymorphism has been shown to be associated with the risk of chronic tactile allodynia after nerve injuries [69]. However, it remains unknown how the disease-predisposing MHCII haplotypes result in the break of immune tolerance to self-antigens, i.e., the production of pathogenic autoimmune Th cells after nerve injuries. The knowledge about central or peripheral immune tolerance and MHCII gene polymorphism in the pathogenesis of organ-specific autoimmune diseases, such as type I diabetes or multiple sclerosis, would provide key paradigms in terms of immunological perspectives and techniques [131, 132]. It is worthy to note, however, that disease-predisposing MHCII haplotypes do not always result in the corresponding autoimmune diseases [62, 69]. In fact, the complex interaction between particular genes, genomic backgrounds, and epigenomic states determines the genetic penetrance of disease-predisposing genes, including MHCII here, in the development of autoimmune diseases [133]. Elucidating this multidimensional interaction in the development of chronic tactile allodynia after nerve injuries will aid in the development of a composite biomarker signature to effectively stratify prospective patients for whom Th cells acts as a trigger for chronic tactile allodynia after nerve injuries [134]. This will facilitate the prevention and treatment of chronic tactile allodynia after nerve injuries by targeting autoimmune Th cells from the perspective of precision medicine.
Conclusions
Elucidating why and how individuals develop or withstand chronic pain after nerve injuries will pave the way for the development of new therapeutic strategies in the context of precision medicine to either prevent or reverse chronic peripheral neuropathic pain. A growing body of clinical and preclinical evidences has clearly showed that Th cells act as an important trigger for chronic tactile allodynia after nerve injuries in some susceptible individuals. Therefore, for translation pain research, an increased intention should be paid to the detailed roles and mechanisms for Th cells in the transition to chronic tactile allodynia after nerve injuries.
Current studies designed by and for neuroscientists themselves have suffered from the neglected and inadequate adoption of immunological perspective, nomenclature, and techniques of Th cells. Here, we introduce a comprehensive interdisciplinary perspective, Th cell neuroimmunology in particular, for neuroscientists who desire deeper insights into pain neuroimmunology. With the state-of-art methods for studying Th cells available in time, future studies with an interdisciplinary perspective will ultimately provide a more comprehensive understanding of whether, where, how, and why Th cells do during the development of chronic tactile allodynia after nerve injuries. This will lay down the foundation for the prevention and treatment of chronic tactile allodynia after nerve injuries by targeting autoimmune Th cells from the perspective of precision medicine.
Availability of data and materials
Not applicable.
Abbreviations
- AICD:
-
Activation-induced cell death
- APC:
-
Antigen-presenting cell
- BBB:
-
Blood-brain barrier
- CCI:
-
chronic constriction injury
- CCK2 :
-
Cholecystokinin type 2 receptor
- CIPN:
-
Chemotherapy-induced peripheral neuropathy
- CNS:
-
Central nervous system
- CPA:
-
Conditioned place aversion
- CRPS:
-
Complex regional pain syndrome
- CSF:
-
Cerebrospinal fluid
- cTEC:
-
Cortical thymic epithelial cell
- CTS:
-
Carpal tunnel syndrome
- DC:
-
Dendritic cell
- DH:
-
Dorsal horn
- DP:
-
Double positive
- DR:
-
Dorsal root
- DRLM:
-
Dorsal root leptomeninge
- EAE:
-
Experimental autoimmune encephalomyelitis
- FRC:
-
Fibroblastic reticular cell
- fTreg:
-
Follicular Treg
- GABA:
-
γ-Aminobutyric acid
- GBS:
-
Guillain-Barre syndrome
- HLA:
-
Human leukocyte antigen
- iTreg:
-
Inducible Treg
- LDH:
-
Lumbar disc herniation
- LLN:
-
Lumbar lymph node
- LTMR:
-
Low-threshold mechanoreceptor
- MCP-3:
-
Monocyte chemotactic protein 3
- MHCII:
-
Major histocompatibility complex class II
- MOR:
-
Mu opioid receptor
- mTEC:
-
Mmedullary thymic epithelial cell
- NK:
-
Natural killer
- NKT:
-
Natural killer T cell
- PDN:
-
Painful diabetic neuropathy
- pDRG:
-
Parenchyma of dorsal root ganglia
- PHN:
-
Postherpetic neuralgia
- PLZF:
-
Promyelocytic leukemia zinc finger
- pMHCII:
-
Peptide-MHCII
- PNS:
-
Peripheral nervous system
- PSN:
-
Primary sensory neuron
- PSNL:
-
Partial sciatic nerve ligation
- PTN:
-
Pain transmission neuron
- PV:
-
Parvalbumin
- RVM:
-
Rostral ventromedial medulla
- SAA:
-
Subarachnoid angle
- SC:
-
Spinal cord
- SC-DH:
-
Spinal cord dorsal horn
- SD:
-
Sprague-Dawley
- SNI:
-
Spared nerve injury
- SP:
-
Single positive
- SSNL:
-
Selective spinal nerve ligation
- TCR:
-
T cell receptor
- TFH:
-
Follicular T helper
- TGN:
-
Trigeminal neuralgia
- Th:
-
T helper
- TLT:
-
Tertiary lymphoid tissue
- TNT:
-
Tibial nerve transection
- TRA:
-
Tissue-restricted antigen
- UEA:
-
Ubiquitous-expressed antigen
- VFH:
-
von Frey hair
References
Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Primers. 2017;3:17002.
Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 2014;13(9):924–35.
Borsook D, Youssef AM, Simons L, Elman I, Eccleston C. When pain gets stuck: the evolution of pain chronification and treatment resistance. Pain. 2018;159(12):2421–36.
Denk F, McMahon SB, Tracey I. Pain vulnerability: a neurobiological perspective. Nat Neurosci. 2014;17(2):192–200.
Price TJ, Basbaum AI, Bresnahan J, Chambers JF, De Koninck Y, Edwards RR, et al. Transition to chronic pain: opportunities for novel therapeutics. Nat Rev Neurosci. 2018;19(7):383–4.
Moehring F, Halder P, Seal RP, Stucky CL. Uncovering the cells and circuits of touch in normal and pathological settings. Neuron. 2018;100(2):349–60.
Tashima R, Koga K, Sekine M, Kanehisa K, Kohro Y, Tominaga K, et al. Optogenetic activation of non-nociceptive Abeta fibers induces neuropathic pain-like sensory and emotional behaviors after nerve injury in rats. eNeuro. 2018;5(1).
Xu ZZ, Kim YH, Bang S, Zhang Y, Berta T, Wang F, et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med. 2015;21(11):1326–31.
Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–9.
Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–7.
Talbot S, Foster SL, Woolf CJ. Neuroimmunity: physiology and pathology. Annu Rev Immunol. 2016;34:421–47.
Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. 2018;100(6):1292–311.
Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7(4):482–93.
O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327(5969):1098–102.
Du B, Ding YQ, **ao X, Ren HY, Su BY, Qi JG. CD4+ αβ T cell infiltration into the leptomeninges of lumbar dorsal roots contributes to the transition from acute to chronic mechanical allodynia after adult rat tibial nerve injuries. J Neuroinflammation. 2018;15(1):81.
Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, et al. Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep. 2016;16(3):605–14.
Tashima R, Mikuriya S, Tomiyama D, Shiratori-Hayashi M, Yamashita T, Kohro Y, et al. Bone marrow-derived cells in the population of spinal microglia after peripheral nerve injury. Sci Rep. 2016;6:23701.
Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27(45):12396–406.
Peng J, Gu N, Zhou L, Be U, Murugan M, Gan WB, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun. 2016;7:12029.
Denk F, Crow M, Didangelos A, Lopes DM, McMahon SB. Persistent alterations in microglial enhancers in a model of chronic pain. Cell Rep. 2016;15(8):1771–81.
Abraira VE, Ginty DD. The sensory neurons of touch. Neuron. 2013;79(4):618–39.
Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, et al. Identification of spinal circuits transmitting and gating mechanical pain. Cell. 2014;159(6):1417–32.
Cheng L, Duan B, Huang T, Zhang Y, Chen Y, Britz O, et al. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat Neurosci. 2017;20(6):804–14.
Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, et al. Dorsal horn circuits for persistent mechanical pain. Neuron. 2015;87(4):797–812.
Koch SC, Acton D, Goulding M. Spinal circuits for touch, pain, and itch. Annu Rev Physiol. 2018;80:189–217.
Petitjean H, Pawlowski SA, Fraine SL, Sharif B, Hamad D, Fatima T, et al. Dorsal horn parvalbumin neurons are gate-keepers of touch-evoked pain after nerve injury. Cell Rep. 2015;13(6):1246–57.
Inquimbert P, Moll M, Latremoliere A, Tong CK, Whang J, Sheehan GF, et al. NMDA receptor activation underlies the loss of spinal dorsal horn neurons and the transition to persistent pain after peripheral nerve injury. Cell Rep. 2018;23(9):2678–89.
Hughes SW, Hickey L, Hulse RP, Lumb BM, Pickering AE. Endogenous analgesic action of the pontospinal noradrenergic system spatially restricts and temporally delays the progression of neuropathic pain following tibial nerve injury. Pain. 2013;154(9):1680–90.
Zhang W, Gardell S, Zhang D, **e JY, Agnes RS, Badghisi H, et al. Neuropathic pain is maintained by brainstem neurons co-expressing opioid and cholecystokinin receptors. Brain. 2009;132(Pt 3):778–87.
Burgess SE, Gardell LR, Ossipov MH, Malan TP Jr, Vanderah TW, Lai J, et al. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22(12):5129–36.
Podvin S, Yaksh T, Hook V. The emerging role of spinal dynorphin in chronic pain: a therapeutic perspective. Annu Rev Pharmacol Toxicol. 2016;56:511–33.
Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013;154(Suppl 1):S10–28.
Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136(Pt 3):828–43.
Ley K. The second touch hypothesis: T cell activation, homing and polarization. F1000Res. 2014;3:37.
Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. 2014;14(6):377–91.
Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol. 2006;6(2):127–35.
Walker LS, Abbas AK. The enemy within: kee** self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2(1):11–9.
Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100.
Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what's in a name? Nat Rev Immunol. 2004;4(3):231–7.
Cohen NR, Brennan PJ, Shay T, Watts GF, Brigl M, Kang J, et al. Shared and distinct transcriptional programs underlie the hybrid nature of iNKT cells. Nat Immunol. 2013;14(1):90–9.
Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, et al. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96(18):10338–43.
Kotov DI, Jenkins MK. Peptide:MHCII tetramer-based cell enrichment for the study of epitope-specific CD4(+) T cells. Curr Protoc Immunol. 2019;125(1):e75.
Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. 2008;9(9):1055–64.
Prince AL, Watkin LB, Yin CC, Selin LK, Kang J, Schwartzberg PL, et al. Innate PLZF+CD4+ αβ T cells develop and expand in the absence of Itk. J Immunol. 2014;193(2):673–87.
Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;29(3):391–403.
Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A. 2014;111(7):2674–9.
Zhang S, Laouar A, Denzin LK, Sant'Angelo DB. Zbtb16 (PLZF) is stably suppressed and not inducible in non-innate T cells via T cell receptor-mediated signaling. Sci Rep. 2015;5:12113.
Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4(8):733–9.
Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37(6):1091–103.
Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13(5):309–20.
Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24(18):4333–9.
Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature. 2016;530(7590):349–53.
Hawse WF, Boggess WC, Morel PA. TCR signal strength regulates Akt substrate specificity to induce alternate murine Th and T regulatory cell differentiation programs. J Immunol. 2017;199(2):589–97.
Turner MS, Isse K, Fischer DK, Turnquist HR, Morel PA. Low TCR signal strength induces combined expansion of Th2 and regulatory T cell populations that protect mice from the development of type 1 diabetes. Diabetologia. 2014;57(7):1428–36.
Stubbington MJT, Rozenblatt-Rosen O, Regev A, Teichmann SA. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017;358(6359):58–63.
Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature. 2019;565(7737):101–5.
Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163(6):1400–12.
Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353(6301):766–71.
Cupovic J, Onder L, Gil-Cruz C, Weiler E, Caviezel-Firner S, Perez-Shibayama C, et al. Central nervous system stromal cells control local CD8(+) T cell responses during virus-induced neuroinflammation. Immunity. 2016;44(3):622–33.
Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, et al. Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity. 2015;43(6):1160–73.
Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462(7269):94–8.
Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6(4):271–82.
King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. 2012;37(4):709–20.
Kotov DI, Mitchell JS, Pengo T, Ruedl C, Way SS, Langlois RA, et al. TCR affinity biases Th cell differentiation by regulating CD25, Eef1e1, and Gbp2. J Immunol. 2019;202(9):2535–45.
Lever M, Maini PK, van der Merwe PA, Dushek O. Phenotypic models of T cell activation. Nat Rev Immunol. 2014;14(9):619–29.
Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210(12):2569–82.
Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol. 2013;4:321.
van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity. 2014;41(1):63–74.
Dominguez CA, Kalliomaki M, Gunnarsson U, Moen A, Sandblom G, Kockum I, et al. The DQB1 *03:02 HLA haplotype is associated with increased risk of chronic pain after inguinal hernia surgery and lumbar disc herniation. Pain. 2013;154(3):427–33.
Dorsey SG, Renn CL, Griffioen M, Lassiter CB, Zhu S, Huot-Creasy H, et al. Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain. PLoS One. 2019;14(5):e0216539.
Tian P, Ma XL, Wang T, Ma JX, Yang X. Correlation between radiculalgia and counts of T lymphocyte subsets in the peripheral blood of patients with lumbar disc herniation. Orthop Surg. 2009;1(4):317–21.
Cheng L, Fan W, Liu B, Wang X, Nie L. Th17 lymphocyte levels are higher in patients with ruptured than non-ruptured lumbar discs, and are correlated with pain intensity. Injury. 2013;44(12):1805–10.
Moalem-Taylor G, Baharuddin B, Bennett B, Krishnan AV, Huynh W, Kiernan MC, et al. Immune dysregulation in patients with carpal tunnel syndrome. Sci Rep. 2017;7(1):8218.
Russo MA, Fiore NT, van Vreden C, Bailey D, Santarelli DM, McGuire HM, et al. Expansion and activation of distinct central memory T lymphocyte subsets in complex regional pain syndrome. J Neuroinflammation. 2019;16(1):63.
Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC. Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation. 2015;12:12.
Kaufmann I, Eisner C, Richter P, Huge V, Beyer A, Chouker A, et al. Lymphocyte subsets and the role of Th1/Th2 balance in stressed chronic pain patients. Neuroimmunomodulat. 2007;14(5):272–80.
Heyn J, Azad SC, Luchting B. Altered regulation of the T-cell system in patients with CRPS. Inflamm Res. 2019;68(1):1–6.
Dominguez CA, Lidman O, Hao JX, Diez M, Tuncel J, Olsson T, et al. Genetic analysis of neuropathic pain-like behavior following peripheral nerve injury suggests a role of the major histocompatibility complex in development of allodynia. Pain. 2008;136(3):313–9.
Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. 2008;38(2):448–58.
Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129(3):767–77.
Zhang X, Wu Z, Hayashi Y, Okada R, Nakanishi H. Peripheral role of cathepsin S in Th1 cell-dependent transition of nerve injury-induced acute pain to a chronic pain state. J Neurosci. 2014;34(8):3013–22.
Makker PGS, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, et al. Characterisation of immune and neuroinflammatory changes associated with chemotherapy-induced peripheral neuropathy. PLoS One. 2017;12(1):e0170814.
Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, et al. CD8(+) T cells and endogenous IL-10 are required for resolution of chemotherapy-induced neuropathic pain. J Neurosci. 2016;36(43):11074–83.
Cobos EJ, Nickerson CA, Gao F, Chandran V, Bravo-Caparros I, Gonzalez-Cano R, et al. Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling. Cell Rep. 2018;22(5):1301–12.
Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29(46):14415–22.
Labuz D, Schreiter A, Schmidt Y, Brack A, Machelska H. T lymphocytes containing β-endorphin ameliorate mechanical hypersensitivity following nerve injury. Brain Behav Immun. 2010;24(7):1045–53.
Vicuna L, Strochlic DE, Latremoliere A, Bali KK, Simonetti M, Husainie D, et al. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat Med. 2015;21(5):518–23.
Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. Cisplatin educates CD8(+) T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain. 2019;160(6):1459–68.
Sweitzer SM, White KA, Dutta C, DeLeo JA. The differential role of spinal MHC class II and cellular adhesion molecules in peripheral inflammatory versus neuropathic pain in rodents. J Neuroimmunol. 2002;125(1–2):82–93.
Fan KQ, Li YY, Wang HL, Mao XT, Guo JX, Wang F, et al. Stress-induced metabolic disorder in peripheral CD4(+) T cells leads to anxiety-like behavior. Cell. 2019;179(4):864.
Huang T, Lin SH, Malewicz NM, Zhang Y, Zhang Y, Goulding M, et al. Identifying the pathways required for co** behaviours associated with sustained pain. Nature. 2019;565(7737):86–90.
Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–3.
Rosen SF, Ham B, Drouin S, Boachie N, Chabot-Dore AJ, Austin JS, et al. T-cell mediation of pregnancy analgesia affecting chronic pain in mice. J Neurosci. 2017;37(41):9819–27.
Echeverry S, Shi XQ, Rivest S, Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci. 2011;31(30):10819–28.
Grace PM, Hutchinson MR, Bishop A, Somogyi AA, Mayrhofer G, Rolan PE. Adoptive transfer of peripheral immune cells potentiates allodynia in a graded chronic constriction injury model of neuropathic pain. Brain Behav Immun. 2011;25(3):503–13.
Hu P, Bembrick AL, Keay KA, McLachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav Immun. 2007;21(5):599–616.
Leger T, Grist J, D'Acquisto F, Clark AK, Malcangio M. Glatiramer acetate attenuates neuropathic allodynia through modulation of adaptive immune cells. J Neuroimmunol. 2011;234(1–2):19–26.
Gattlen C, Clarke CB, Piller N, Kirschmann G, Pertin M, Decosterd I, et al. Spinal cord T-cell infiltration in the rat spared nerve Iinjury model: a time course study. Int J Mol Sci. 2016;17(3):352.
Bali KK, Kuner R. Therapeutic potential for leukocyte elastase in chronic pain states harboring a neuropathic component. Pain. 2017;158(11):2243–58.
Kim CF, Moalem-Taylor G. Detailed characterization of neuro-immune responses following neuropathic injury in mice. Brain Res. 2011;1405:95–108.
Lopes DM, Malek N, Edye M, Jager SB, McMurray S, McMahon SB, et al. Sex differences in peripheral not central immune responses to pain-inducing injury. Sci Rep. 2017;7(1):16460.
Austin PJ, Kim CF, Perera CJ, Moalem-Taylor G. Regulatory T cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain. 2012;153(9):1916–31.
Perera CJ, Duffy SS, Lees JG, Kim CF, Cameron B, Apostolopoulos V, et al. Active immunization with myelin-derived altered peptide ligand reduces mechanical pain hypersensitivity following peripheral nerve injury. J Neuroinflammation. 2015;12:28.
Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, et al. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A (3) adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun. 2015;44:91–9.
Austin PJ, Berglund AM, Siu S, Fiore NT, Gerke-Duncan MB, Ollerenshaw SL, et al. Evidence for a distinct neuro-immune signature in rats that develop behavioural disability after nerve injury. J Neuroinflammation. 2015;12:96.
Galbavy W, Kaczocha M, Puopolo M, Liu L, Rebecchi MJ. Neuroimmune and neuropathic responses of spinal cord and dorsal root ganglia in middle age. PLoS One. 2015;10(8):e0134394.
Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–31.
Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112(1):23–38.
McKelvey R, Berta T, Old E, Ji RR, Fitzgerald M. Neuropathic pain is constitutively suppressed in early life by anti-inflammatory neuroimmune regulation. J Neurosci. 2015;35(2):457–66.
McLachlan EM, Hu P. Inflammation in dorsal root ganglia after peripheral nerve injury: effects of the sympathetic innervation. Auton Neurosci. 2014;182:108–17.
Schmid AB, Coppieters MW, Ruitenberg MJ, McLachlan EM. Local and remote immune-mediated inflammation after mild peripheral nerve compression in rats. J Neuropathol Exp Neurol. 2013;72(7):662–80.
Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain. 2011;12(3):370–83.
Younger DS, Rosoklija G, Hays AP, Trojaborg W, Latov N. Diabetic peripheral neuropathy: a clinicopathologic and immunohistochemical analysis of sural nerve biopsies. Muscle Nerve. 1996;19(6):722–7.
Moalem G, Monsonego A, Shani Y, Cohen IR, Schwartz M. Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J. 1999;13(10):1207–17.
Hartlehnert M, Derksen A, Hagenacker T, Kindermann D, Schafers M, Pawlak M, et al. Schwann cells promote post-traumatic nerve inflammation and neuropathic pain through MHC class II. Sci Rep. 2017;7(1):12518.
Kobayashi Y, Kiguchi N, Fukazawa Y, Saika F, Maeda T, Kishioka S. Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J Biol Chem. 2015;290(20):12603–13.
Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85(3):493–502.
Liu XJ, Zhang YL, Liu T, Xu ZZ, Park CK, Berta T, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014;24(11):1374–7.
Agarwal N, Helmstadter J, Rojas DR, Bali KK, Gangadharan V, Kuner R. Evoked hypoalgesia is accompanied by tonic pain and immune cell infiltration in the dorsal root ganglia at late stages of diabetic neuropathy in mice. Mol Pain. 2018;14:1744806918817975.
Carow B, Gao Y, Coquet J, Reilly M, Rottenberg ME. Lck-driven Cre expression alters T cell development in the thymus and the frequencies and functions of peripheral T cell subsets. J Immunol. 2016;197(6):2261–8.
Baron EM. Spinal cord and spinal nerves: gross anatomy. In: Standring S, editor. Gray’s anatomy: the anatomical basis of clinical practice. 41st ed. Singapore: Elsevier; 2015. p. 762–73.
Joukal M, Klusakova I, Dubovy P. Direct communication of the spinal subarachnoid space with the rat dorsal root ganglia. Ann Anat. 2016;205:9–15.
Braun JS, Kaissling B, Le Hir M, Zenker W. Cellular components of the immune barrier in the spinal meninges and dorsal root ganglia of the normal rat: immunohistochemical (MHC class II) and electron-microscopic observations. Cell Tissue Res. 1993;273(2):209–17.
Zenker W, Bankoul S, Braun JS. Morphological indications for considerable diffuse reabsorption of cerebrospinal fluid in spinal meninges particularly in the areas of meningeal funnels. An electronmicroscopical study including tracing experiments in rats. Anat Embryol (Berl). 1994;189(3):243–58.
Reina MA, De Leon Casasola Ode L, Villanueva MC, Lopez A, Maches F, De Andres JA. Ultrastructural findings in human spinal pia mater in relation to subarachnoid anesthesia. Anesth Analg. 2004;98(5):1479–85. table of contents
Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82(4):981–1011.
Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Forstera B, Zhao S, et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull-meninges connections. Nat Neurosci. 2019;22(2):317–27.
**g D, Zhang S, Luo W, Gao X, Men Y, Ma C, et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res. 2018;28(8):803–18.
Wang Q, Zhang S, Liu T, Wang H, Liu K, Wang Q, et al. Sarm1/Myd88-5 regulates neuronal intrinsic immune response to traumatic axonal injuries. Cell Rep. 2018;23(3):716–24.
Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. 2015;125(2):699–714.
Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-l-fucose synthase is a CD4(+) T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci Transl Med. 2018;10(462).
van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes. 2014;63(1):237–47.
Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132(10):1077–130.
Tracey I, Woolf CJ, Andrews NA. Composite pain biomarker signatures for objective assessment and effective treatment. Neuron. 2019;101(5):783–800.
Acknowledgements
Not applicable.
Funding
The work reported herein was supported by grants from the National Science Foundation of China (31571240) to Jian-Guo Qi, Sichuan Province Development and Regeneration Key Laboratory Program (SYS17-007) to You-Quan Ding, and China Postdoctoral Science Foundation (2019 M653416) and Post-Doctor Research Project, West China Hospital, Sichuan University (2018HXBH004) to Han Luo.
Author information
Authors and Affiliations
Contributions
YQD conceived and designed the framework of the review, performed literature researching and sorting, analyzed most of the data, prepared the figures, and wrote the manuscript. HL gave important suggestions to the framework of the review, performed some literature researching and sorting, analyzed some data, and revised the manuscript, figures, and tables. JGQ conceived and designed the framework of the review, supervised the execution of this project, and contributed to data interpretation and manuscript preparation. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Funding institutions have no role in study design, data collection, data analysis and interpretation, article writing, or submitting decision for publication. The authors declare no conflicts of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ding, YQ., Luo, H. & Qi, JG. MHCII-restricted T helper cells: an emerging trigger for chronic tactile allodynia after nerve injuries. J Neuroinflammation 17, 3 (2020). https://doi.org/10.1186/s12974-019-1684-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1684-0