
Abstract
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a critical tumor suppressor protein that regulates various biological processes such as cell proliferation, apoptosis, and inflammatory responses by controlling the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PI3K/AKT) signaling pathway. PTEN plays a crucial role in the pathogenesis of rheumatoid arthritis (RA). Loss of PTEN may contribute to survival, proliferation, and pro-inflammatory cytokine release of fibroblast-like synoviocytes (FLS). Also, persistent PI3K signaling increases myeloid cells’ osteoclastic potential, enhancing localized bone destruction. Recent studies have shown that the expression of PTEN protein in the synovial lining of RA patients with aggressive FLS is minimal. Experimental upregulation of PTEN protein expression could reduce the damage caused by RA. Nonetheless, a complete comprehension of aberrant PTEN drives RA progression and its interactions with other crucial molecules remains elusive. This review is dedicated to promoting a thorough understanding of the signaling mechanisms of aberrant PTEN in RA and aims to furnish pertinent theoretical support for forthcoming endeavors in both basic and clinical research within this domain.
Similar content being viewed by others
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder that targets joints and is characterized by persistent synovial inflammation, proliferation of synovial tissue, synovial lichen planus, and subsequent damage to nearby articular cartilage and bone [1]. It impacts around 1% of the world’s population [2]. Typically diagnosed between the ages of 40 and 50, this condition is more common in females, with a three to five times greater occurrence rate compared to males [3]. The main clinical presentation involves repeated and symmetrical episodes of polyarthritis affecting several joints such as the hands, wrists, feet, and knees. Typical symptoms at the first stages include redness, swelling, fever, discomfort, and joint dysfunction [4]. The pathophysiology of RA is sophisticated and complex, and it is widely considered that several variables, including genetic and environmental factors, synergistically disrupt the immune system, resulting to an aberrant immunological response. Moreover, immune and non-immune cells work together to sustain chronic inflammation, resulting in decreased activity and eventually diminished quality of life [5]. In the initial phase of the disease, the immune system becomes active, leading to the infiltration of various inflammatory cells such as mast cells, macrophages, monocytes, fibroblasts, and chondrocyte-like cells into the synovial tissues of the affected joints. As the infiltration rises, RA gets progressively more severe and the synovial tissue is severely infiltrated by leukocytes, which finally leads to proliferation of the endothelium layer [6, 7]. Patients typically show increased levels of rheumatoid factor (RF), anti-citrullinated protein/peptide antibodies (ACPA), and other distinctive indicators [8].New pathways in the pathogenesis of RA include dendritic cell-T cell interaction, pyroptosis, and autophagy, which have been prominent in recent years [9, 10]. The primary therapeutic goals for RA involve pain relief, inflammation control, immune response suppression, cartilage protection, and disease progression delay [11]. Commonly prescribed medications include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying antirheumatic drugs (DMARDs). Despite the availability of various treatment options, a significant number of RA patients do not achieve remission [12]. Signal transduction mechanisms are crucial in the development of RA, with abnormal activation of signaling pathways leading to the production of inflammatory molecules [13, 14]. As a result, identifying effective molecular targets for RA treatment is a key focus of research.
PI3K pathway governs fundamental processes such as cell survival, migration, proliferation, cytoskeletal remodeling, and immune cell homeostasis [15]. PTEN, an antagonistic phosphatase of various PI3Ks, ranks as the second most commonly mutated gene in human cancers after p53, indicating prognostic assessment in numerous tumors [16]. Beyond tumors, PTEN plays a pivotal role in various chronic inflammatory diseases, including chronic nephritis, gastritis, and RA. FLS constitute a significant cell population within the invasive pannus and function as crucial mediators of the local inflammatory response in RA [17]. Recent findings suggest that PTEN deficiency can amplify the survival and growth of FLS in RA, potentially due to PTEN’s regulatory role in the secretion and release of inflammatory factors associated with RA-FLS, including interleukin (IL)-6/1β, chemokine (C-C motif) ligand (CCL)-23, and vascular endothelial growth factor (VEGF) [18, 19]. Preliminary studies have explored the connection between PTEN and RA in the literature, however the precise processes responsible for the abnormal expression of PTEN in RA and its association with RA inflammation and FLS activation are not yet fully understood. We will review recent literature on the molecular structure of PTEN and its regulatory mechanisms. We will then discuss the evidence supporting PTEN as a potential target in RA, highlighting its involvement in regulating RA-FLS inflammation, joint damage in RA, and angiogenesis in RA. The goal is to thoroughly assess the existing knowledge and pinpoint areas where comprehension is lacking.
Structure of PTEN
PTEN is made up of 403 amino acids and has three functional domains (Fig. 1): the N-terminal [PI(4,5)P2]-binding/phosphatase domain, C2 domain, and carboxyl-terminal tail (C-tail) domain. The N-terminal domain of PTEN, namely residues 15–186, serves as the catalytic core, containing the HCxxGxxR active site within the P-loop (amino acids 123–130). Cancer-related missense mutations tend to accumulate in this specific area [20, 21]. The N-terminal phosphatase and C-terminal C2 structural domains play crucial roles in PTEN’s catalytic activity and intracellular distribution. The C-terminal C2 domain has structural features that enable attachment to the phospholipid bilayer, even though it does not have the loop structure for Ca2+ binding and Ca2+-dependent membrane contact [22, 23]. Additionally, N-terminal residues 6 to 15 participate in membrane interactions, particularly with the phospholipid PIP2, underscoring their importance in PTEN’s cellular function of down-regulating PI3K signaling [24]. The C-terminal region of PTEN contains identifiable motifs such as potential phosphorylation sites, a PDZ-binding motif, and two PEST motifs. Phosphorylating or dephosphorylating the serine and threonine residues at the C-terminal is crucial for maintaining the stability, positioning, and enzymatic function of PTEN [25, 26]. The PDZ-binding domain mostly interacts with particular cell surface receptors or intracellular proteins, aiding in PTEN’s localization to specific cellular areas [27, 28]. The PEST sequence functions as an intracellular marker for the half-life of proteins undergoing proteolytic degradation. Deletion of this region typically leads to an increase in PTEN’s expression [29]. The segment is not required for ubiquitin-ligase NEDD4-1 to ubiquitinate PTEN [30]. Further investigation is required to determine the roles of these two PEST sequences.

PTEN structures. A: Overall view of the PTEN crystal structure. The cartoon representation of the PTEN crystal structure (PDB ID: 7jvx) displays the phosphatase domain in pink and the C2 structural domain in blue-green. B: The domain architecture of PTEN. PTEN consists of 403 amino acids, and the protein has three functional domains: the N-terminal domain which binds and phosphatizes PI(4,5)P2, the C2 domain, and the carboxy-terminal tail domain (C-tail). The catalytically active site on the P-loop at the bottom of the active site pocket: HCxxGxxR (amino acids 123–130, red). The C2 domain plays a key role in membrane binding. PTEN also contains two PEST structural domains and a PDZ structural domain, PDZ binding domains can localize PTEN to specific cellular regions, and PEST sequences may be correlated with PTEN expression. there is a set of phosphorylation sites at the C-terminus that play an important role in maintaining PTEN stability and catalytic activity
PTEN was first identified as a suppressor of the PI3K pathway in the cytoplasm, but it has also been found in the nucleus, where it can act as a tumor suppressor by regulating the cell cycle and maintaining genome stability, separate from its role in the PI3K pathway [31]. The labeling of cytoplasmic proteins into the nucleus is called nuclear localization signal (NLS), and most of the initially reported NLSs are abundant in basic amino acids, hence this form of NLS is named basic NLS or classical NLS (cNLS) [32]. These are classified into two distinct types: monopartite and bipartite. The former is a simple sequence consisting of 4–8 residues, like the first identified SV40 big T antigen NLS (PKKKRKV); the latter is two basic sequences with a separation of approximately 10–12 residues [33]. Various methods of PTEN transportation into the nucleus have been identified. Initially, PTEN moves through nuclear pore complexes (NPCs) to reach the nucleus [34]; Subsequently, the transport receptor Importin-11(IPO11) transfers PTEN to the nucleus and inhibits its breakdown in the cytoplasm. Importin-β (IPO1) and Transportin-2 (TNPO2 or IPO3) are two proteins that facilitate transport [35, 36]. Additionally, oxidative stress hinders the nuclear export of PTEN to safeguard cells and control tumorigenesis. The inhibitory impact was determined to be associated with the phosphorylation of the amino acid residue Ser380 [37].
Notably, PTEN mRNA can undergo alternative translation initiation to generate multiple long isoforms of PTEN with additional N-terminal extensions. The initial ones identified were PTENα (576 amino acids)and PTENβ (549 amino acids) [38]. PTENα utilizes the same mRNA as PTEN, but it initiates translation from the non-AUG start codon CUG and includes an ATR region at the N-terminus [39]. PTENα is a membrane-permeable lipid phosphatase that is secreted by cells and can enter other cells to function as a secreted PI3K antagonist. PTENα is situated on the outer mitochondrial membrane and enhances mitochondrial autophagy by aiding in the recruitment of Parkinson disease protein 2(PARK2) to impaired mitochondria [40]. Moreover, research discovered that transcription activator-like effector nucleases (TALENs) -induced removal of PTENα in somatic cells hinders the operation of the mitochondrial respiratory chain. The translation of PTENβ is reported to be in-frame with the AUG start sequence of a conventional PTEN. PTENβ is mostly found in the nucleolus, where it directly interacts with and removes phosphate groups from nuciferin. Disruption of PTENβ results in an abnormal rise in pre-rRNA production and encourages cell growth [41]. Aside from PTENα and PTENβ, there are a minimum of four other N-terminally elongated versions of PTEN that can be produced by utilizing the start codon located in the 5′UTR region of the PTEN mRNA [42]. Recent investigations have shown the existence of PTENε (475 amino acids), whose natural reduction leads to the development of filamentous pseudopods and boosts the spreading ability of cancer cells [42].
PTEN functions
PTEN stands as the inaugural tumor suppressor gene identified to possess dual phosphatase characteristics for both lipid and protein phosphatases [43]. Operating within the cytoplasm and cell membrane, PTEN governs pivotal cellular processes, including cell survival, proliferation, senescence, and angiogenesis (Fig. 2). This regulation occurs predominantly through inhibiting the PI3K-AKT pathway, primarily executed via lipophosphatase activity [44, 45]. Upon reception of external signals, such as growth factors or insulin, cells activate PI3K, leading to the generation of PIP3. PIP3, in turn, binds to AKT, initiating AKT phosphorylation expression and impeding apoptotic signaling pathways, thereby fostering cell survival [46]. Furthermore, AKT can indirectly stimulate proteins associated with the mechanistic target of rapamycin (mTOR). The activation of mTOR assumes a pivotal role in diverse cellular activities, encompassing protein synthesis, autophagy, nucleotide synthesis, and cell growth, achieved through the phosphorylation of various downstream effector proteins [43]. PTEN, functioning as a critical pathway regulator, effectively thwarts AKT activation by dephosphorylating PIP3 and converting it to PIP2. Consequently, this process sustains in vivo homeostasis [47].

Illustrates the functions of PTEN in the cytoplasm and nucleus. A: Persistent activation of the PI3K/AKT signaling pathway and subsequent mTOR activation upon PTEN loss. This cascade, initiated by the phosphorylation of downstream effector proteins, contributes to processes such as protein and nucleotide synthesis. B: PTEN interacts with MVP in the nucleus in a Ca + dependent way and has a significant function in preserving genome stability while interacting with various proteins
PTEN is primarily known as a lipid phosphatase that hinders the activation of the oncogenic PI3K-AKT signaling pathway. PTEN has additional potential modes of action, such as protein phosphatase activity. Qi et al. showed that the protein phosphatase function of PTEN is necessary for epithelial differentiation and polarization in the epiblast [48]. Furthermore, PTEN controls cell cycle and death by utilizing its protein phosphatase activity, performing a similar function to AKT [49]. Xu et al. discovered that PTEN’s protein phosphatase activity dephosphorylates and inhibits autophosphorylated phosphoglycerate kinase 1 (PGK1), leading to the inhibition of glycolysis, ATP synthesis, and brain tumor cell proliferation [50]. In addition, Helen and colleagues utilized the mutant enzyme PTENY138L, which specifically lacks protein phosphatase activity, to study PTEN in transgenic mice lacking either all PTEN function or only protein phosphatase activity in the prostate gland. They discovered that PTEN protein phosphatase activity was not essential for tumor suppression [51]. PTENα deficiency enhances mitochondrial autophagy triggered by different mitochondrial-damaging substances. A laboratory test showed that PTENα removes phosphate groups from pSer65-Ub using its protein phosphatase activity, regardless of its lipid phosphatase function [39]. PTEN plays crucial roles in the nucleus by regulating chromosome integrity, chromatin structure, DNA replication, and damage repair [52]. PTEN gains access into the nucleus through a calcium-dependent pathway by interacting with the main vault protein (MVP) [34]. Subsequently, genome stability is maintained via its interaction with CENP-C, a centromere-specific binding protein, which is essential for ensuring centromere stability. PTEN also has a role in regulating the expression of RAD51, a key protein involved in repairing DNA damage from double-stranded breaks (DSBs) [53, 54]. Moreover, PTEN is crucial in DNA replication since it interacts with many replication-related proteins such as replication protein A1 (RPA1) and microchromosome maintenance complex component 2 (MCM2) at DNA replication forks [55]. A study revealed that the activation of the G2/M checkpoint, triggered by the DNA damage response (DDR), involves the engagement of p53 and PTEN in NSCLC cells using a dynamic Boolean network and experimental data [56].
Mechanisms of PTEN activity regulation
The loss of PTEN function typically results in the accumulation of PIP3, subsequently activating the AKT signaling pathway. Additionally, PTEN deficiency is associated with the activation of multiple pathways through PIP3-dependent signaling, including the Ras-MAPK, Wnt/β-catenin, Notch, and Hippo pathways [43]. Predominant efforts to reinstate PTEN loss have predominantly focused on inhibiting PI3K. However, a substantial proportion of disorders, encompassing tumors, exhibit impairment in PTEN activation [57]. As depicted in Table 1, various regulatory mechanisms influence PTEN activity, including transcriptional regulation (involving miRNAs) and post-transcriptional translational modifications such as phosphorylation, ubiquitination, acetylation, and methylation [58].
Transcriptional regulation
Regulating PTEN levels includes several methods. RNA-binding motif protein 24 (RBM24) binds directly to PTEN mRNA during the transcription step, thereby stabilizing it [64]. The work further shows that miR-214 promotes the AKT signaling pathway by directly targeting PTEN and MIR31HG. Disrupting the MIR31HG-miR-214-PTEN pathway inhibits the growth, movement, and production of inflammatory factors and MMPs in RA-FLS [65]. miR-3-1p, activated by specificity protein 1(SP1), enhances chondrocyte pyroptosis triggered by IL-1β by suppressing PTEN and activating the PINK1/Parkin pathway [66]. This discovery offers new perspectives on investigating miRNA-induced localized cell death and the possible pathways involved in the development of rheumatoid arthritis. MiR-10b disturbs the equilibrium of CD4 T cell subsets, leading to the progression of rheumatoid arthritis. PTEN was discovered as a target of miR-10b. Using PTEN siRNA increased Th17 cells and decreased Treg cells [67]. When the AKT/mTOR pathway is inhibited, miRNAs significantly downregulate the expression of phosphatase and PTEN [68].
Post-translational modifier
PTEN activity is regulated through several changes, including ubiquitination, phosphorylation, acetylation, and methylation. Here, we provide a summary of recent discoveries in this field. HMG-CoA reductase degradation protein 1(HRD1), Syntaxin3(STX3), and Glutamine–Fructose-6-Phosphate Transaminase 1(GFPT1) have been recognized as facilitators of PTEN degradation through ubiquitination, resulting in enhanced cancer cell proliferation [69,70,71]. Nuclear PTEN is more likely to be deleted than cytoplasmic PTEN, as it is less stable and more prone to degradation through the ubiquitin-proteasome pathway [72]. Lysine 13 ubiquitination controls the nuclear localization of PTEN, preventing DNA damage in living organisms [73]. PTEN degradation occurs due to the ubiquitination of lysine 221, leading to a decrease in RAD51 expression and causing instability in double-strand breaks (DSB) by hindering homologous recombination (HR) [74]. Phosphorylation of PTEN at tyrosine 240 boosts DNA repair mechanisms and provides protection against DNA damage. Crucially, the distinct phosphorylation action of PTEN functions independently of its lipid phosphatase activity [75]. ATM phosphorylates PTEN at serine 113, promoting the movement of PTEN into the nucleus and triggering autophagy [76]. The activation of the S-phase checkpoint is dependent on the phosphorylation of PTEN at position 398, which is regulated by PI3K-p27 [77]. Acetylation of lysine 402 on PTEN may regulate its interaction with proteins that have the PDZ structural domain [78]. Deacetylating PTEN hinders the ubiquitination of epidermal growth factor receptor (EGFR), which strengthens extracellular matrix formation, triggers autophagy in chondrocytes, and offers defense against osteoarthritis [79].
DNA methylation is a crucial epigenetic alteration in animals that has important impacts on the growth, development, and progression of diseases, particularly in cancer research. Changes in DNA methylation in RA-FLS occur early in the progression of RA, before a confirmed clinical diagnosis is made. DNA methylation is suggested as a possible main cause of RA persistence [80]. A study found a link between the immunological response in the blood of people with active RA and changes in DNA methylation patterns of circulating monocytes. The study found many clusters of CpG sites with methylation levels closely linked to the disease activity ratings of individuals [81]. Recent research has revealed a significant link between tumors and autoimmune illnesses, emphasizing hypermethylation as a route for PTEN inactivation. Li and colleagues suggested that the decrease in PTEN expression may be associated with CpG site methylation. This was supported by the discovery of four CpG sites near and before the first exon of the transcript that had a high concentration of C and G nucleotides. The methylation inhibitor 5-Azacytidine significantly increased PTEN expression in adjuvant-induced arthritis (AIA) and reduced the mRNA and protein levels of Tumor necrosis factor alpha (TNF-α), Interleukin 6 (IL-6), and IL-1β [82]. Recent research highlights the influence of PTEN methylation on inflammation and activation in FLS. Preventing PTEN methylation led to increased amounts of anti-inflammatory molecules and decreased activation of FLS in people with RA [83]. DNMTs catalyze the onset of DNA methylation, and their inhibition in experimental arthritis angiogenesis suppresses DNMT1-mediated PTEN hypermethylation [84].
PTEN as an emerging target in RA
Involved in RA-FLS inflammatory regulation
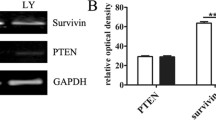
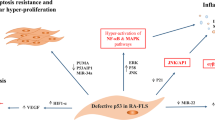
RA-FLS, specialized mesenchymal cells, are found in the synovium of both joints. RA causes FLS to display many biological characteristics such as aberrant growth, increased aggressiveness, and excessive secretion of inflammatory substances and cytokines, including TNF-α, IL-6, C-C chemokine ligand(CCL-2), Matrix metalloproteinase-3(MMP3), and vascular endothelial growth factor-alpha (VEGF-α). These processes lead to excessive synovial tissue formation and eventual joint damage [85, 86]. The molecular pathways responsible for activated FLS proliferation and migration are not well understood, however PTEN may play a role in this complex pathway [19]. Studies show that the absence of PTEN results in the creation of substances that cause arthritis and degeneration of the joints [87]. Increasing PTEN expression leads to alterations in the PI3K/AKT signaling pathway, resulting in a delay in RA progression [88]. Reducing PTEN expression using either the PTEN inhibitor BPV or PTEN-RNAi was found to increase the growth and movement of FLS [19] (Fig. 3B). NFIC, a part of the NFI family, acts as a transcription factor that boosts PTEN transcription. This process results in RA-FLS cell death and successfully decreases inflammation in mice with collagen-induced arthritis (CIA) [89]. Glutathione (GSH), an antioxidant, reduces inflammation in FLS by adjusting pathways associated with PTEN [90]. Boosting PTEN levels in inflamed tissues using recombinant adenoviruses or decreasing PTEN phosphorylation markedly reduced the inflammatory reaction by inhibiting AKT [88, 91]. PTEN overexpression reduced T-cell activation and influenced the development of Th17 and Treg cells, improving experimental autoimmune arthritis. The progression of autoimmune arthritis is hindered by the inadequate levels of PTEN in myeloid cells, which limits the development of harmful Th17-type immune responses [22, 92]. The PI3K/PTEN pathway is suggested to regulate various aspects of disease development in inflammatory conditions in different tissues, including cell movement, invasiveness, cytokine generation, cell growth, and T-cell orientation. The results enhance comprehension of regulatory complexities, as seen in Table 2.

Description of PTEN as an emerging target in RA. A: PTEN deficiency promotes VECs movement and infiltration; delayed pericyte development; and inhibition of VSMCs differentiation, ultimately leading to an imbalance in vascular homeostasis. B: PTEN deficiency promotes the proliferation and invasion of FLS and the formation of a proactive inflammatory relief, leading to excessive synovial tissue formation and ultimately joint injury. C: PTEN deficiency disrupts the balance between osteoblasts and osteoclasts, promotes osteoclast survival, decreases chondrocyte viability, and accelerates bone degeneration
Involved in the destruction of bone and cartilage in RA joints
RA is defined by inflammation that results in the deterioration of bone and cartilage in the joints it affects. RA triggers an inflammatory response that leads to bone loss by increasing osteoclastic bone resorption and reducing osteoblastic bone growth [93, 94]. This imbalance results in bone degradation, loss of bone around the joints, and widespread osteoporosis. Osteoclasts play a crucial role in bone degradation as shown in basic research and the efficacy of antibody treatments produced by osteoclasts in medical practice [95]. Osteoclasts originate from hematopoietic stem cells and differentiate within the monocyte-macrophage lineage from osteoclast precursors [96]. Recent research indicates that PI3K has a role in osteoclast formation, and activating the PI3K/AKT/mTOR pathway can enhance osteoblast development. Specific PI3K/AKT inhibitors, including LY294002 and LY3023414, have shown the capacity to decrease bone growth in both living organisms and laboratory settings [97, 98]. PTEN loss has been linked to increased Early growth response factor 1 (EGR1) expression, which plays a role in controlling osteoclast formation and encouraging metastasis [99]. Friedrich’s research suggests that removing PTEN and maintaining the lack of PI3K signaling in myeloid cells can enhance osteoclastogenesis in myeloid cells, leading to localized bone degradation [100]. Liu et al. used Cre-mediated recombination to selectively disrupt the PTEN in osteoblasts. Osteoblasts lacking PTEN developed faster, had lower apoptosis rates, and showed a notable rise in phosphorylated AKT levels compared to the control samples [101].
Two vital cytokines, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-kappa B ligand (RANKL), are essential for the differentiation of osteoclasts [102, 103]. RANKL, a cytokine attached to the TNF superfamily and cell membrane, interacts with receptor activator of nuclear factor-kappa B (RANK)on osteoclast precursors, triggering osteoclast development by activating nuclear factor of activated T-cells 1 (NFATc1) as a key regulator. RANKL affects PTEN in two ways in the control of bone homeostasis (Fig. 3B). Initially, it suppresses the expression and function of PTEN phosphatase, promoting cell survival and growth. Concurrently, RANKL stimulation triggers the PI3K/AKT cascade, resulting in the deactivation of PTEN and Glycogen synthase kinase 3 Beta (GSK3β), ultimately facilitating osteoclast formation [104]. PTEN loss boosts the final development of RANKL-induced osteoclasts via elevating NFATc1 expression, as demonstrated in in vitro experiments [105]. PTEN has been discovered to interact with miRNAs that control the survival of mature osteoblasts. Enhancing mitochondrial autophagy in osteoclast precursors hinders osteoclast development and decreases bone damage in collagen-induced arthritic mice. The PINK1/Parkin pathway mainly controls mitochondrial autophagy [106]. Notably, PTEN has two distinct roles. Its overexpression in degenerating disc cells leads to apoptosis in these cells, reducing chondrocyte viability and inhibiting the formation of type II collagen by blocking PI3K/AKT activation [107].
Involved in angiogenesis in RA
Angiogenesis is crucial for promoting effective bone regeneration by allowing the transfer of nutrients, growth factors, and waste products, which helps maintain the internal balance of the skeleton [108]. Excessive angiogenic factors in RA counteract anti-angiogenic factors, resulting in increased endothelial cell infiltration, heightened synovial inflammation, and eventual bone and cartilage destruction. Inhibiting angiogenesis in the joint has the potential to reduce synovitis and limit the growth of sub-synovial tissue [86, 109]. Targeting angiogenesis could be a crucial strategy in treating RA [110]. Various pro-inflammatory mediators involved in stimulating the formation of new blood vessels in rheumatoid arthritis are being assessed as possible targets for upcoming treatments. These consist of chemokines (CXCL12), cytokines (IL-17, IL-18, and MIF), growth factors (Ang1 and Ang2), proteases (MMPs), and adhesion molecules (ICAM1 and VCAM1) [111]. Approved treatments for rheumatoid arthritis that may work by inhibiting angiogenesis include TNF, IL-1β, and IL-6 inhibitors, thalidomide, and Cox-2 inhibitors.
PTEN’s involvement in RA angiogenesis is becoming more acknowledged f(Fig. 3A). Suppressing PTEN expression has been demonstrated to enhance the growth, movement, infiltration, and formation of new blood vessels by vascular endothelial cells (VECs) [112]. Increased PTEN expression boosts the biological activities of HUVECs, resulting in a suppressive impact on angiogenesis [113]. Intravascular homeostasis is maintained through the cooperation of endothelial cells, pericytes, and vascular smooth muscle cells (vSMCs), which play a crucial role in stabilizing and controlling vascular function. Pericyte deficiency is linked to several disorders such as diabetic retinopathy and cancer [114]. PTEN loss causes a delay in pericyte development, leading to the activation of PI3K signaling. Pericyte maturation is necessary for vascular remodeling during angiogenesis [13]. Increased expression of PTEN reduces angiotensin II-induced damage while also decreasing fibrosis and inflammatory indicators [115]. 5-Azacytidine, a DNMT1 inhibitor, has been found to enhance PTEN expression, promote the preservation of smooth muscle cell differentiation, and reduce pathological vascular remodeling [116]. Depletion of PTEN in mice leads to prolonged PI3K-AKT-mTOR signaling, resulting in decreased expression of vSMC markers. The decrease occurs simultaneously with the activation of NF-κβ and the production of chemokines and substances that promote fibrosis (MCP-1, IL-6, and KC/CXCL1), aiding in injury-induced vascular adjustment [117]. Interestingly, PTEN loss was associated with neointima development, leading to the increased expression of genes associated with inflammation and fibrosis [118]. Stromal cell-derived factor-1 alpha (SDF-1α) acts as an intermediary following PTEN. When PTEN is lost in vascular smooth muscle cells, it causes an upregulation of SDF-1α expression, leading to the emergence of an inflammatory phenotype characterized by the attraction of bone marrow-derived progenitor cells [119]. Moreover, important targets further along the PTEN pathway have been associated with abnormal vascular adaptation. The findings highlight the significant role of PTEN signaling in rheumatoid arthritis angiogenesis, involving endothelial cells and all components of the arterial wall structure [120].
Discussion and outlook
RA is a prevalent autoimmune inflammatory condition. If left untreated, persistent inflammation of the joint lining can result in significant joint deterioration, disability, and inability to function [121]. PTEN is the initial oncogene identified to possess bispecific phosphatase activity, and is a gene that is highly linked to cancer following the p53. The quantity of articles on PTEN is growing annually. PTEN in the cytoplasm is known to operate as a negative regulator of the PI3K pathway, influencing the RA inflammatory response, bone degradation, and angiogenesis. The decrease of PTEN activity occurs through various routes. At the transcriptional stage, PTEN is silenced by epigenetic mechanisms by the targeted control of numerous proteins. Recent research has shown that inhibiting the PIP3 phosphatase activity of PTEN can be achieved by targeting the PTEN catalytic core (aa118-141) [122]. PTEN activity is controlled through ubiquitination, phosphorylation, acetylation, and methylation changes in the translational phase. DNA methylation is being viewed as a potential target for therapeutic and diagnostic purposes in RA. The transcription products of PTEN contain numerous CpG sites, therefore, the main attention should be on the effects of PTEN hypermethylation on the disease. PTEN is present in the nucleus and has a role in regulating DNA damage, maintaining genomic stability, inhibiting oncogenic transcription, and is not dependent on the PI3K pathway. Deletion of nuclear PTEN is linked to many cancer characteristics. Post-translational changes are essential for the nuclear translocation and stability of PTEN. Ultimately, the cause of RA is intricate, and examining the PTEN’s upstream and downstream actions can provide valuable understanding of gene regulation’s complexity and diversity.
An urgent need exists in the clinical field for new antirheumatic drugs that show increased effectiveness [123]. Current research suggests that using PTEN as a therapeutic target for RA is still in the early stages. Illustrative methods involve using adenoviral or genetically modified bionic membrane-encapsulated vectors for mRNA therapies, employing herbal extracts to adjust the molecular mechanisms related to the anti-inflammatory or immunomodulatory effects of PTEN, and using methylation inhibitors or antioxidants to alter pathways associated with PTEN [88, 124, 125]. Among them, Traditional Chinese remedies provide beneficial therapeutic effects on RA, offering a greater number of targets and less adverse effects compared to conventional therapeutic pharmaceuticals. They are a key area of interest in RA drug development research. Guizhi Shaoyao Zhimu granules (GSZGs) enhance the process of autophagy in mitochondria of osteoclast precursors through the PTEN-induced PINK1/Parkin pathway, resulting in reduced bone degradation in mice with CIA [106]. Catalpol is a bioactive compound derived from the traditional herb Radix Rehmanniae. Catalpol increases PTEN function by reducing PTEN ubiquitination and degradation, which leads to the inhibition of the RANKL-induced NF-κB and AKT signaling pathways. It shows promise for treating RA and other bone-related disorders [126].Overall, Strategies for one-way control of PTEN expression levels should be further explored in future medication development. RA herbal formulae derived from traditional chinese medicine (TCM) clinical practice require additional testing in vivo and in vitro. It is essential to produce active components with reduced side effects and broader target coverage in collaboration with pharmacology. Future research should concentrate on investigating the role of PTEN in the progression of RA to enhance comprehension and offer direction for the diagnosis and treatment of RA.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- PI3K/AKT:
-
phosphatidylinositol 3-kinase (PI3K)/protein kinase B
- RA:
-
rheumatoid arthritis
- FLS:
-
fibroblast-like synoviocytes
- RF:
-
rheumatoid factor
- ACPA:
-
anti-citrullinated protein/peptide antibodies
- NSAIDs:
-
nonsteroidal anti-inflammatory drugs
- DMARDs:
-
disease-modifying antirheumatic drugs
- VEGF:
-
vascular endothelial growth factor
- C-tail:
-
carboxyl-terminal tail
- NLS:
-
nuclear localization signal
- cNLS:
-
classical NLS
- NPCs:
-
nuclear pore complexes
- IPO1:
-
Importin-β
- IPO11:
-
Importin-11
- TNPO2:
-
Transportin-2
- TALENs:
-
transcription activator-like effector nucleases
- PGK1:
-
phosphoglycerate kinase 1
- MVP:
-
main vault protein
- DSBs:
-
double-stranded breaks
- RPA1:
-
replication protein A1
- MCM2:
-
maintenance complex component 2
- DDR:
-
DNA damage response
- RBM24:
-
RNA-binding motif protein 24
- EZH2:
-
Enhancer of Zeste Homolog 2
- miRNAs:
-
MicroRNAs
- STAT3:
-
signal transducer and activator of transcription 3
- PRDM1:
-
PR domain zinc finger protein 1
- HRD1:
-
HMG-CoA reductase degradation protein 1
- STX3:
-
Syntaxin3
- GFPT1:
-
Glutamine–Fructose-6-Phosphate Transaminase 1
- DSB:
-
double-strand breaks
- HR:
-
homologous recombination
- EGFR:
-
epidermal growth factor receptor
- AIA:
-
adjuvant-induced arthritis
- TNF-α:
-
Tumor necrosis factor alpha
- IL-6:
-
Interleukin 6
- CCL-2:
-
C-C chemokine ligand
- MMP3:
-
Matrix metalloproteinase-3
- VEGF-α:
-
vascular endothelial growth factor-alpha
- CIA:
-
collagen-induced arthritis
- GSH:
-
Glutathione
- M-CSF:
-
macrophage colony-stimulating factor
- RANKL:
-
receptor activator of nuclear factor-kappa B ligand
- RANK:
-
receptor activator of nuclear factor-kappaB
- NFATc1:
-
nuclear factor of activated T-cells 1
- GSK3β:
-
Glycogen synthase kinase 3 Beta
- VECs:
-
vascular endothelial cells
- vSMCs:
-
vascular smooth muscle cells
- GSZGs:
-
Guizhi Shaoyao Zhimu granules
- TCM:
-
Traditional Chinese Medicine
References
Langbour C, Rene J, Goupille P, et al. Fficacy of Janus kinase inhibitors in rheumatoid arthritis [J]. Inflamm Res. 2023;72:1121–32.
Korol I, Baumeister RH. Treating rheumatoid arthritis [J]. JAAPA. 2023;36(9):1–5.
Watanabe R, Hashimoto M, Murata K, et al. Prevalence and predictive factors of difficult-to-treat rheumatoid arthritis [J]: the KURAMA cohort. Immunol Med. 2022;45(1):35–44.
Ding Q, Hu W, Wang R et al. Signaling pathways in rheumatoid arthritis: implications for targeted therapy [J]. Signal Transduct Target Therapy. 2023;8.
Radu AF, Bungau SG. Nanomedical approaches in the realm of rheumatoid arthritis [J]. Ageing Res Rev. 2023;87:101927.
Meng M, Yao J, Zhang Y, et al. Potential anti-rheumatoid arthritis activities and mechanisms of Ganoderma Lucidum polysaccharides [J]. Molecules. 2023;28:2483.
Zhu M, Ding Q, Lin Z, et al. New targets and strategies for rheumatoid arthritis: from Signal Transduction to Epigenetic aspect [J]. Biomolecules. 2023;13:766.
Shams S, Martinez JM, Dawson JRD, et al. The Therapeutic Landscape of Rheumatoid Arthritis: current state and future directions [J]. Front Pharmacol. 2021;12:680043.
Wang Z, Zhang J, An F, et al. The mechanism of dendritic cell-T cell crosstalk in rheumatoid arthritis [J]. Arthritis Res Ther. 2023;25(1):193.
Wu D, Li Y, Xu R. Can pyroptosis be a new target in rheumatoid arthritis treatment? [J]. Front Immunol. 2023;14:1155606.
Garaffoni C, Adinolfi A, Bortoluzzi A, et al. Novel insights into the management of rheumatoid arthritis: one year in review 2023 [J]. Clin Exp Rheumatol. 2023;41(11):2129–41.
Venetsanopoulou AI, Voulgari PV, Drosos AA. Advances in non-biological drugs for the treatment of rheumatoid arthritis [J]. Expert Opin Pharmacother. 2024;25(1):45–53.
Figueiredo AM, Villacampa P, Diéguez-Hurtado R, et al. Phosphoinositide 3-Kinase–regulated pericyte maturation governs vascular remodeling [J]. Circulation. 2020;142:688–704.
Philippou E, Petersson SD, Rodomar C, et al. Rheumatoid arthritis and dietary interventions: systematic review of clinical trials [J]. Nutr Rev. 2020;79:410–28.
Banham-Hall E. The therapeutic potential for PI3K inhibitors in Autoimmune Rheumatic diseases [J]. Open Rheumatol J. 2012;6:245–58.
Liu A, Zhu Y, Chen W, et al. PTEN Dual lipid- and protein-phosphatase function in Tumor progression [J]. Cancers. 2022;14:3666.
Radu AF, Bungau SG. Management of rheumatoid arthritis: an overview [J]. Cells. 2021;10(11):2857.
Tong Y, Li X, Deng Q, et al. Advances of the small molecule drugs regulating fibroblast-like synovial proliferation for rheumatoid arthritis [J]. Front Pharmacol. 2023;14:1230293.
Li R, Wu X, Peng S, et al. CCR2 antagonist represses fibroblast-like synoviocyte-mediated inflammation in patients with rheumatoid arthritis [J]. Int Immunopharmacol. 2023;122:110570.
Masson GR, Williams RL. Structural mechanisms of PTEN regulation [J]. Cold Spring Harb Perspect Med. 2020;10(3):a036152.
Tu T, Chen J, Chen L, et al. Dual-specific protein and Lipid Phosphatase PTEN and its biological functions [J]. Cold Spring Harb Perspect Med. 2020;10(1):a036301.
Torices L, Nunes-Xavier CE, López JI, et al. Novel anti-PTEN C2 domain monoclonal antibodies to analyse the expression and function of PTEN isoform variants [J]. PLoS ONE. 2023;18(8):e0289369.
Karn R, Emerson IA. Molecular dynamic study on PTEN frameshift mutations in breast cancer provide c2 domain as a potential biomarker [J]. J Biomol Struct Dyn. 2022;40(7):3132–43.
Henager SH, Henriquez S, Dempsey DR, et al. Analysis of site-specific phosphorylation of PTEN by using enzyme-catalyzed expressed protein ligation [J]. ChemBioChem. 2020;21(1–2):64–8.
Sotelo NS, Schepens JT, Valiente M, et al. PTEN-PDZ domain interactions: binding of PTEN to PDZ domains of PTPN13 [J]. Methods. 2015;77–78:147–56.
Leoni G, Alam A, Neumann PA, et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair [J]. J Clin Invest. 2013;123(1):443–54.
Yan M, Wang Y, Wong CW, et al. PTEN PDZ-binding domain suppresses mammary carcinogenesis in the MMTV-PyMT breast cancer model [J]. Cancer Lett. 2018;430:67–78.
Adey NB, Huang L, Ormonde PA, et al. Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding [J]. Cancer Res. 2000;60(1):35–7.
Georgescu M-M, Kirsch KH, Akagi T, et al. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region [J]. Proc Natl Acad Sci. 1999;96:10182–7.
Dai C, Wu B, Chen Y, et al. Aagab acts as a novel regulator of NEDD4-1-mediated Pten nuclear translocation to promote neurological recovery following hypoxic-ischemic brain damage [J]. Cell Death Differ. 2021;28(8):2367–84.
Langdon CG. Nuclear PTEN’s functions in suppressing tumorigenesis: implications for rare cancers [J]. Biomolecules. 2023;13:259.
Moraes IR, de Oliveira HC, Fontes MRM. Structural basis of nuclear transport for NEIL DNA glycosylases mediated by importin-alpha [J]. Biochim Biophys Acta Proteins Proteom. 2024;1872(2):140974.
Lu J, Wu T, Zhang B et al. Types of nuclear localization signals and mechanisms of protein import into the nucleus [J]. Cell Communication Signal. 2021;19.
Zhang W, Neo SP, Gunaratne J, et al. Feedback regulation on PTEN/AKT pathway by the ER stress kinase PERK mediated by interaction with the Vault complex [J]. Cell Signal. 2015;27(3):436–42.
Bao W, Florea L, Wu N, et al. Loss of nuclear PTEN in HCV-infected human hepatocytes [J]. Infect Agent Cancer. 2014;9:23.
Leslie NR. Importin-11 keeps PTEN safe from harm. J Cell Biol. 2017;216(3):539–41.
Chakraborty S, Karmakar S, Basu M, et al. The E3 ubiquitin ligase CHIP drives monoubiquitylation-mediated nuclear import of the tumor suppressor PTEN [J]. J Cell Sci. 2023;136(18):jcs260950.
Torices L, Nunes-Xavier CE, López JI, et al. Novel anti-PTEN C2 domain monoclonal antibodies to analyse the expression and function of PTEN isoform variants [J]. PLoS ONE. 2023;18:e0289369.
Sun Y, Lu D, Yin Y, et al. PTENα functions as an immune suppressor and promotes immune resistance in PTEN-mutant cancer [J]. Nat Commun. 2021;12(1):5147.
Han R, Liu Y, Li S, et al. PINK1-PRKN mediated mitophagy: differences between in vitro and in vivo models [J]. Autophagy. 2023;19(5):1396–405.
Liang H, He S, Yang J, et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism [J]. Cell Metab. 2014;19(5):836–48.
Zhang Q, Liang H, Zhao X, et al. PTENε suppresses tumor metastasis through regulation of filopodia formation [J]. EMBO J. 2021;40(10):e105806.
Chow JT, Salmena L. Recent advances in PTEN signalling axes in cancer [J]. Fac Rev. 2020;9:31.
Hashemi M, Etemad S, Rezaei S, et al. Progress in targeting PTEN/PI3K/Akt axis in glioblastoma therapy: revisiting molecular interactions [J]. Biomed Pharmacother. 2023;158:114204.
Chen M, Choi S, Wen T, et al. A p53-phosphoinositide signalosome regulates nuclear AKT activation [J]. Nat Cell Biol. 2022;24(7):1099–113.
Huang J, Qi Z. MiR-21 mediates the protection of kaempferol against hypoxia/reoxygenation-induced cardiomyocyte injury via promoting Notch1/PTEN/AKT signaling pathway [J]. PLoS ONE. 2020;15(11):e0241007.
Ashrafizadeh M, Zarrabi A, Samarghandian S, et al. PTEN: what we know of the function and regulation of this onco-suppressor factor in bladder cancer? [J]. Eur J Pharmacol. 2020;881:173226.
Qi Y, Liu J, Chao J et al. PTEN dephosphorylates Abi1 to promote epithelial morphogenesis [J]. J Cell Biology. 2020;219.
Tu T, Chen J, Chen L, et al. Dual-specific protein and Lipid Phosphatase PTEN and its biological functions [J]. Cold Spring Harbor Perspect Med. 2019;10:a036301.
Qian X, Li X, Shi Z, et al. PTEN suppresses glycolysis by Dephosphorylating and Inhibiting Autophosphorylated PGK1 [J]. Mol Cell. 2019;76:516–e5277.
Wise HM, Harris A, Kriplani N, et al. PTEN protein phosphatase activity is not required for Tumour suppression in the mouse prostate [J]. Biomolecules. 2022;12:1511.
Zhang J, Lee YR, Dang F, et al. PTEN methylation by NSD2 Controls Cellular sensitivity to DNA damage [J]. Cancer Discov. 2019;9(9):1306–23.
Zhao L, Li R, Qiu JZ, et al. YY1-mediated PTEN dephosphorylation antagonizes IR-induced DNA repair contributing to tongue squamous cell carcinoma radiosensitization [J]. Mol Cell Probes. 2020;53:101577.
Shen WH, Balajee AS, Wang J, et al. Essential role for Nuclear PTEN in maintaining Chromosomal Integrity [J]. Cell. 2007;128:157–70.
Wang G, Li Y, Wang P, et al. PTEN regulates RPA1 and protects DNA replication forks [J]. Cell Res. 2015;25(11):1189–204.
Gupta S, Panda PK, Silveira DA, et al. Quadra-stable dynamics of p53 and PTEN in the DNA damage response [J]. Cells. 2023;12:1085.
Ha SE, Kim SM, Vetrivel P, et al. Inhibition of cell proliferation and metastasis by Scutellarein regulating PI3K/Akt/NF-κB signaling through PTEN activation in Hepatocellular Carcinoma [J]. Int J Mol Sci. 2021;22(16):8841.
González-García A, Garrido A, Carrera AC. Targeting PTEN Regulation by Post translational modifications [J]. Cancers. 2022;14:5613.
**a RM, Liu T, Li WG, Xu XQ. RNA-binding protein RBM24 represses colorectal tumourigenesis by stabilising PTEN mRNA. Clin Transl Med. 2021;11(10):e383.
Su Y, Wang B, Huang J, et al. YTHDC1 positively regulates PTEN expression and plays a critical role in cisplatin resistance of bladder cancer [J]. Cell Prolif. 2023;56(7):e13404.
Zang X, Gu J, Zhang J, et al. Exosome-transmitted lncRNA UFC1 promotes non-small-cell lung cancer progression by EZH2-mediated epigenetic silencing of PTEN expression [J]. Cell Death Dis. 2020;11(4):215.
Azari H, Nazari E, Mohit R, et al. Machine learning algorithms reveal potential miRNAs biomarkers in gastric cancer [J]. Sci Rep. 2023;13(1):6147.
Heinicke F, Zhong X, Flåm ST, et al. MicroRNA expression differences in blood-derived CD19 + B cells of Methotrexate treated rheumatoid arthritis patients [J]. Front Immunol. 2021;12:663736.
Wan L, Wang Y, Li J, et al. Inhibition of the AKT/mTOR pathway negatively regulates PTEN expression via miRNAs [J]. Acta Biochim Biophys Sin (Shanghai). 2022;54(11):1637–47.
Cao L, Jiang H, Yang J, et al. LncRNA MIR31HG is induced by tocilizumab and ameliorates rheumatoid arthritis fibroblast-like synoviocyte-mediated inflammation via mir-214-PTEN-AKT signaling pathway [J]. Aging. 2021;13(21):24071–85.
Jiang J-M, Mo M-L, Long X-P, et al. MiR-144-3p induced by SP1 promotes IL-1β-induced pyroptosis in chondrocytes via PTEN/PINK1/Parkin axis [J]. Autoimmunity. 2021;55:21–31.
Tu J, Han D, Fang Y, et al. MicroRNA-10b promotes arthritis development by disrupting CD4 + T cell subtypes [J]. Mol Therapy - Nucleic Acids. 2022;27:733–50.
Wan L, Wang Y, Li J, et al. Inhibition of the AKT/mTOR pathway negatively regulates PTEN expression via miRNAs [J]. Acta Biochim Biophys Sin. 2022;54:1637–47.
Li D, Guan M, Cao X, et al. GFPT1 promotes the proliferation of cervical cancer via regulating the ubiquitination and degradation of PTEN [J]. Carcinogenesis. 2022;43:969–79.
Li P, Song R, Yin F, et al. circMRPS35 promotes malignant progression and cisplatin resistance in hepatocellular carcinoma [J]. Mol Ther. 2022;30(1):431–47.
Liu L, Long H, Wu Y, et al. HRD1-mediated PTEN degradation promotes cell proliferation and hepatocellular carcinoma progression [J]. Cell Signal. 2018;50:90–9.
Ge MK, Zhang N, **a L, et al. FBXO22 degrades nuclear PTEN to promote tumorigenesis [J]. Nat Commun. 2020;11(1):1720.
Kato T, Yamada T, Nakamura H, et al. The loss of Nuclear PTEN increases Tumorigenesis in a Preclinical Mouse Model for Hepatocellular Carcinoma [J]. iScience. 2020;23:101548.
Sinha A, Saleh A, Endersby R, et al. RAD51-Mediated DNA homologous recombination is Independent of PTEN Mutational Status [J]. Cancers. 2020;12:3178.
Lin J, Song T, Li C, Mao W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res. 2020;1867(5):118659.
Liu W, Xu L, Wang X, et al. PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage [J]. Autophagy. 2021;17(12):4159–81.
Bassi C, Fortin J, Snow BE, et al. The PTEN and ATM axis controls the G1/S cell cycle checkpoint and tumorigenesis in HER2-positive breast cancer [J]. Cell Death Differ. 2021;28:3036–51.
Lu Q, Liu P, Miao Z, et al. SIRT1 restoration enhances chondrocyte autophagy in osteoarthritis through PTEN-mediated EGFR ubiquitination [J]. Cell Death Discov. 2022;8(1):203.
Huang J, Ye Z, Wang J, Chen Q, Huang D, Liu H. USP13 mediates PTEN to ameliorate osteoarthritis by restraining oxidative stress, apoptosis and inflammation via AKT-dependent manner [J]. Biomed Pharmacother. 2021;133:111089.
Lin X, Tao C, Zhang R, et al. N6-methyladenosine modification of TGM2 mRNA contributes to the inhibitory activity of sarsasapogenin in rheumatoid arthritis fibroblast-like synoviocytes [J]. Phytomedicine. 2022;95:153871.
Julià A, Gómez A, López-Lasanta M, et al. Longitudinal analysis of blood DNA methylation identifies mechanisms of response to tumor necrosis factor inhibitor therapy in rheumatoid arthritis [J]. EBioMedicine. 2022;80:104053.
Guan H, Zhu N, Tang G, et al. DNA methyltransferase 1 knockdown reverses PTEN and VDR by mediating demethylation of promoter and protects against renal injuries in hepatitis B virus-associated glomerulonephritis [J]. Cell Biosci. 2022;12(1):98.
Li XF, Wu S, Yan Q, et al. PTEN Methylation Promotes Inflammation and activation of Fibroblast-Like synoviocytes in Rheumatoid Arthritis [J]. Front Pharmacol. 2021;12:700373.
Bu Y, Wu H, Deng R, et al. Geniposide restricts angiogenesis in experimentary arthritis via inhibiting Dnmt1-mediated PTEN hypermethylation [J]. Int Immunopharmacol. 2022;111:109087.
Chen W, Fang Y, Wang H, et al. Role of chemokine receptor 2 in rheumatoid arthritis: a research update [J]. Int Immunopharmacol. 2023;116:109755.
Meyer A, Zack SR, Nijim W, et al. Metabolic reprogramming by Syntenin-1 directs RA FLS and endothelial cell-mediated inflammation and angiogenesis [J]. Cell Mol Immunol. 2024;21(1):33–46.
Cui X, Liu X, Kong P, et al. PTEN inhibitor VO-OHpic protects endplate chondrocytes against apoptosis and calcification via activating Nrf-2 signaling pathway [J]. Aging. 2023;15(6):2275–92.
Wu S, Wang J, Li J, et al. microRNA-21 aggravates Lipopolysaccharide-Induced inflammation in MH7A cells through targeting SNF5 [J]. Inflammation. 2020;43(2):441–54.
Jia P, Zhang W, Shi Y. NFIC attenuates rheumatoid arthritis-induced inflammatory response in mice by regulating PTEN/SENP8 transcription [J]. Tissue Cell. 2023;81:102013.
Hao WT, Huang L, Pan W, et al. Antioxidant glutathione inhibits inflammation in synovial fibroblasts via PTEN/PI3K/AKT pathway: an in vitro study [J]. Archives Rheumatol. 2021;37:212–22.
Li X, Shi Z, Zhu Y, et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice [J]. Br J Pharmacol. 2020;177(15):3591–607.
Blüml S, Sahin E, Saferding V, et al. Phosphatase and tensin homolog (PTEN) in antigen-presenting cells controls Th17-mediated autoimmune arthritis [J]. Arthritis Res Ther. 2015;17(1):230.
Kitaura H, Marahleh A, Ohori F, et al. Osteocyte-related cytokines regulate osteoclast formation and bone resorption [J]. Int J Mol Sci. 2020;21:5169.
Auréal M, Machuca-Gayet I, Coury F. Rheumatoid arthritis in the view of osteoimmunology [J]. Biomolecules. 2020;11:48.
Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis — immune cell–fibroblast–bone interactions [J]. Nat Reviews Rheumatol. 2022;18:415–29.
Udagawa N, Koide M, Nakamura M, et al. Osteoclast differentiation by RANKL and OPG signaling pathways [J]. J Bone Min Metab. 2021;39(1):19–26.
Sun K, Luo J, Guo J, et al. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: a narrative review [J]. Osteoarthr Cartil. 2020;28:400–9.
Chen X, Chen W, Aung ZM, et al. LY3023414 inhibits both osteogenesis and osteoclastogenesis through the PI3K/Akt/GSK3 signalling pathway [J]. Bone Joint Res. 2021;10(4):237–49.
You J, Wu Q, Li Y, et al. Lentinan induces apoptosis of mouse hepatocellular carcinoma cells through the EGR1/PTEN/AKT signaling axis [J]. Oncol Rep. 2023;50(1):142.
Blüml S, Friedrich M, Lohmeyer T, et al. Loss of phosphatase and tensin homolog (PTEN) in myeloid cells controls inflammatory bone destruction by regulating the osteoclastogenic potential of myeloid cells [J]. Ann Rheum Dis. 2013;74:227–33.
Liu X, Bruxvoort KJ, Zylstra CR, et al. Lifelong accumulation of bone in mice lacking PTEN osteoblasts [J]. Proc Natl Acad Sci. 2007;104:2259–64.
Souza PPC, Lerner UH. Finding a toll on the Route: the Fate of Osteoclast progenitors after Toll-Like receptor activation [J]. Front Immunol. 2019;10:1663.
Kim J-M, Lin C, Stavre Z, et al. Osteoblast-Osteoclast Communication Bone Homeost [J] Cells. 2020;9:2073.
Lu SH, Hsia YJ, Shih KC, et al. Fucoidan prevents RANKL-Stimulated osteoclastogenesis and LPS-Induced inflammatory bone loss via regulation of Akt/GSK3β/PTEN/NFATc1 signaling pathway and calcineurin activity [J]. Mar Drugs. 2019;17(6):345.
Chen H, Li S, Yin H, et al. MYC-mediated miR-320a affects receptor activator of nuclear factor κB ligand (RANKL)-induced osteoclast formation by regulating phosphatase and tensin homolog (PTEN) [J]. Bioengineered. 2021;12(2):12677–87.
Yao H, **ang L, Huang Y, et al. Guizhi Shaoyao Zhimu granules attenuate bone destruction in mice with collagen-induced arthritis by promoting mitophagy of osteoclast precursors to inhibit osteoclastogenesis [J]. Phytomedicine. 2023;118:154967.
Cui X, Liu X, Kong P, et al. PTEN inhibitor VO-OHpic protects endplate chondrocytes against apoptosis and calcification via activating Nrf-2 signaling pathway [J]. Aging. 2023;15:2275–92.
Zhao F, He Y, Zhao Z, et al. The notch signaling-regulated angiogenesis in rheumatoid arthritis: pathogenic mechanisms and therapeutic potentials [J]. Front Immunol. 2023;14:1272133.
Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes [J]. Nat Rev Rheumatol. 2020;16(6):316–33.
Liu C, He L, Wang J, et al. Anti-angiogenic effect of Shikonin in rheumatoid arthritis by downregulating PI3K/AKT and MAPKs signaling pathways [J]. J Ethnopharmacol. 2020;260:113039.
Wang Y, Wu H, Deng R. Angiogenesis as a potential treatment strategy for rheumatoid arthritis [J]. Eur J Pharmacol. 2021;910:174500.
Jiang F, Wang MQ, Zhang MY, et al. CPD-002, a novel VEGFR2 inhibitor, relieves rheumatoid arthritis by reducing angiogenesis through the suppression of the VEGFR2/PI3K/AKT signaling pathway [J]. Int Immunopharmacol. 2024;131:111850.
Bu Y, Wu H, Deng R, et al. The anti-angiogenesis mechanism of Geniposide on rheumatoid arthritis is related to the regulation of PTEN [J]. Inflammopharmacology. 2022;30:1047–62.
van Splunder H, Villacampa P, Martínez-Romero A, et al. [J]. Pericytes in the disease spotlight. Trends Cell Biol. 2024;34(1):58–71.
Lu S, Strand KA, Mutryn MF, et al. PTEN (phosphatase and Tensin Homolog) protects against Ang II (angiotensin II)-Induced pathological vascular fibrosis and remodeling-brief report [J]. Arterioscler Thromb Vasc Biol. 2020;40(2):394–403.
Strand KA, Lu S, Mutryn MF, et al. High throughput screen identifies the DNMT1 (DNA Methyltransferase-1) inhibitor, 5-Azacytidine, as a potent inducer of PTEN (phosphatase and Tensin Homolog): Central Role for PTEN in 5-Azacytidine Protection Against pathological vascular remodeling [J]. Arterioscler Thromb Vasc Biol. 2020;40(8):1854–69.
Fan L, Liu C, Chen X, et al. Exosomes-Loaded Electroconductive Hydrogel synergistically promotes tissue repair after spinal cord Injury via Immunoregulation and Enhancement of Myelinated Axon growth [J]. Adv Sci (Weinh). 2022;9(13):e2105586.
Furgeson SB, Simpson PA, Park I, et al. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation [J]. Cardiovasc Res. 2010;86(2):274–82.
Nemenoff RA, Horita H, Ostriker AC, et al. SDF-1α induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation [J]. Arterioscler Thromb Vasc Biol. 2011;31(6):1300–8.
Orozco-García E, van Meurs DJ, Calderón J, et al. Endothelial plasticity across PTEN and Hippo pathways: a complex hormetic rheostat modulated by extracellular vesicles [J]. Translational Oncol. 2023;31:101633.
Brown P, Pratt AG, Hyrich KL. Therapeutic advances in rheumatoid arthritis [J]. BMJ. 2024;e070856.
Shang M, Ni L, Shan X, et al. MTHFD2 reprograms macrophage polarization by inhibiting PTEN [J]. Cell Rep. 2023;42:112481.
Meehan RT, Amigues IA, Knight V. Precision Medicine for Rheumatoid Arthritis: the right drug for the Right Patient-Companion Diagnostics [J]. Diagnostics (Basel). 2021;11(8):1362.
Chen J, Tan J, Li J et al. Genetically Engineered Biomimetic nanoparticles for targeted delivery of mRNA to treat rheumatoid arthritis [J]. Small Methods. 2023.
Almansour ZH, Ibrahim H-IM, Hamad RS, et al. Phenolic-compound-rich Opuntia littoralis Ethyl acetate Extract relaxes arthritic symptoms in Collagen-Induced mice Model via Bone morphogenic markers [J]. Nutrients. 2022;14:5366.
Meng J, Zhang W, Wang C, et al. Catalpol suppresses osteoclastogenesis and attenuates osteoclast-derived bone resorption by modulating PTEN activity [J]. Biochem Pharmacol. 2020;171:113715.
Acknowledgements
Not applicable.
Funding
This study was supported in part by the Special Project for Traditional Chinese Medicine Research of Sichuan Provincial Administration of Traditional Chinese Medicine (2020LC0010).
Author information
Authors and Affiliations
Contributions
Writing—review and editing: Pan Zhou, **ngwen Meng; data collection: ZhiminNie; conceptualization: Hua Wang; funding acquisition: Aihua Du; Kaijun Wang administration: Yu Lei;
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, P., Meng, X., Nie, Z. et al. PTEN: an emerging target in rheumatoid arthritis?. Cell Commun Signal 22, 246 (2024). https://doi.org/10.1186/s12964-024-01618-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01618-6