
Abstract
The molecules of Major histocompatibility class I (MHC-I) load peptides and present them on the cell surface, which provided the immune system with the signal to detect and eliminate the infected or cancerous cells. In the context of cancer, owing to the crucial immune-regulatory roles played by MHC-I molecules, the abnormal modulation of MHC-I expression and function could be hijacked by tumor cells to escape the immune surveillance and attack, thereby promoting tumoral progression and impairing the efficacy of cancer immunotherapy. Here we reviewed and discussed the recent studies and discoveries related to the MHC-I molecules and their multidirectional functions in the development of cancer, mainly focusing on the interactions between MHC-I and the multiple participators in the tumor microenvironment and highlighting the significance of targeting MHC-I for optimizing the efficacy of cancer immunotherapy and a deeper understanding of the dynamic nature and functioning mechanism of MHC-I in cancer.
Similar content being viewed by others


Introduction
The cells from jawed vertebrates need to provide the immune system with the physiological status information, thus inducing necessary elimination in face of infection and magnificent transformation or maintaining the immune tolerance under normal conditions [1, 2]. This process is dominated by the molecules of Major histocompatibility class I (MHC-I) complex, which presents the peptides on the surface of the cell. The MHC-I complex displays the structure of heterodimers, consisting the polymorphic heavy chain and the light chain β2-microglobulin (B2M) [3,4,5]. After presenting the peptides on the cell surface, the MHC-I complexes would get scanned by the T-cell receptors (TCRs), allowing the CD8+ T cells to recognize the antigenic peptides and then clean the cancerous or infected cells [6]. Obviously, this process serves as the primary step and a fundamental basis for the anti-cancer immunity and accordingly, any interference or abnormal regulation during this process may be hijacked by tumor cells to escape the immune surveillance and elimination.
Recent years witnessed great advances made in cancer immunotherapy, especially immune-checkpoint blockade (ICB), cancer vaccines, and chimeric antigen receptor (CAR) T-cells, striving to reinvigorate the T cell-mediated anti-cancer immunity to kill the cancer cells. Although significant survival benefits have been brought to a wide range of cancer patients, great difficulty and barrier in the treatment still exist, including the innate and adaptive immune resistance, complex tumor immune microenvironment, great individual difference, and the difficulty in predicting the immunotherapy effect. These challenges still seriously limited the clinical application of cancer immunotherapy.
As we mentioned before, MHC-I-mediated antigenic peptides presentation pathway is the predominate initiative factor for the anti-cancer immunity. Therefore, the importance of the MHC-I modulation in cancer immune evasion has been emphasized in recent years and a range of studies have reported that the loss or downregulation of MHC-I is a major mechanism of cancer immune evasion by blocking the surface presentation of tumor-associated antigens, thus suppressing the cytotoxicity of CD8+ T cells and impairing the adaptive immune response [7, 8]. Nevertheless, in addition to this obvious logical mechanism, multiple studies also uncovered a range of non-canonical biological functions of MHC-I molecules in cancer, posing a totally different direction in multiple aspects, including the partner immune cell subtypes, immune functioning mechanisms, and the interactive relationships with the tumor microenvironment. Thus, we came to realize that MHC-I molecules could participate in the tumorigenesis through multiple pathways. Meanwhile, the emerging newly-identified roles played by MHC-I molecules in the tumor microenvironment pose new questions and challenges to us. Firstly, since the MHC-I could engage in the immune response regulation of various immune cells, are these processes carried out simultaneously and independently or coupling with each other? Secondly, what up-stream factors determine or influence the direction of the MHC-I molecule-mediated tumor immunity and how the positive and negative immune effects caused by MHC-I achieve the mutual balance and transformation? Moreover, how can we utilize the mechanism of MHC-I dysfunction in anti-cancer immunity to develop more effective diagnostic and therapeutic approaches to battling against cancer immune evasion and invigorating the cancer immunotherapy efficacy? To answer these questions, we are required to comprehensively review the key studies and related literature of MHC-I in cancer context and further clarify the complex networks centered on MHC-I in the tumor microenvironment. As the first step, we need to recognize and understand the structural basis of MHC-I.
The structure and construction of MHC-I molecules
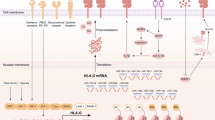
As the material basis of adaptive immune system, the evolutionary inception of MHC complexes (MHC-I and MHC-II) dates back about 500 million years ago [9]. MHC-I molecules are commonly located on the cell surface of the nucleated cells, forming the trimeric complexes composed of a heavy chain, an invariant light chain B2M. The heavy chains are lined with the cell membrane and the domains distant to the membrane, including α1 and α2, form a groove structure for the peptide binding. The heavy chains of MHC-I are known as Human Leukocyte Antigen (HLA), which are genetically encoded by HLA-A, HLA-B, and HLA-C as the classical MHC-I genes and HLA-E, HLA-F, and HLA-G as the non-classical MHC-I genes. High polymorphism exists within the MHC-I genes results in different allelic variants of the MHC-I molecules, thus ensuring the great diversity of the bound peptide ligands and the uniqueness of the distinct presented peptide repertoires [10]. Also, the binding of the peptide endows the MHC-I complex with stabilization. Once the MHC-I molecule is loaded with the peptides, the MHC-I complex will become peptide/MHC-I (pMHC-I). The peptide displayed determines how CD8+ T cells will treat the pMHC-I expressed cells. When the peptides loaded by MHC-I are “non-self” with abnormally altered structures, the CD8+ T cells would be activated following the antigen recognition and then induce the immune killing to the aberrantly antigen-exposed cells. Meanwhile, for the pMHC-I complexes with “self” peptides, tonic signals would be released for the survival of the naïve CD8+ T cells [11].
The pathway of the assembly and construction of pMHC-I begins in the endoplasmic reticulum (ER). Firstly, both the classical and non-classical neoantigens would be packaged into the proteosomes in the cytosol to get trimmed into neoantigen peptides. Then the Transporter associated with Antigen Processing (TAP) will transfer the peptides to the ER, where the peptides will be further trimmed into smaller and optimized sizes (8–10 amino acids) through the aminopeptidase function of the ER aminopeptidases (ERAP)1 and ERAP2. Calnexin, a molecular chaperone of MHC-I, plays a fundamental role in the folding and assembly MHC-I heavy chain, forming the partially folded MHC-I. Afterwards, the peptide-loading complex, constructed by the TAP subunits TAP1 and TAP2, calreticulin, ERp57, and tapasin, will get the peptide loaded on the MHC-I. Subsequently, the pMHC-I molecules are transferred to the plasma membrane to be presented to the CD8+ T cells. Any defect in the production or function of the components of MHC-I, such as the HLA heavy chains, B2M light chains, or the peptides production and loading complex subunits, etc., will interfere the normal presentation of MHC-I molecules on the cell surface and subsequently affect the immune recognition and response of CD8+ T cells.
Regulation of MHC-I expression
As an immune protein complex, the expression of MHC-I molecule is modulated by multiple mechanisms. Diverse levels could be utilized to regulate the expression of MHC-I, which provides a finer and more complex regulatory network. At the level of genetic transcription, the major regulator for the MHC-I molecule genes is Nucleotide-binding oligomerization domain-Like Receptor family Caspase recruitment domain containing 5 (NLRC5), also known as the Class I Transactivator (CITA). By binding with RFX5 and RFXAP, NLRC5 forms a CITA enhanceosome complex to promote the expression of MHC-I molecules as a transcriptional activator. As a member of leucine-rich containing proteins (NLR) family, NLRC5 also represents a major sensor for recognizing endogenous and exogenous stress and microbial infection to boost innate and adaptive immunity. The classical cellular functions of NLRC5 have been nicely reviewed and summarized by a range of studies [12]. As an upstream factor, IFN molecules (IFN-α, β, and γ) increase the expression of MHC-I through the JAK/STAT pathway, which subsequently activate the transcriptional functions of NF-κB and IRF to bind to the Enhancer A region and the interferon-stimulated response element (ISRE) in the promoter region of MHC-I and thus promote the expression of MHC-I [13, 14]. Notably, a range of studies also revealed that NLRC5 could also in turn modulate the IFN responses in different ways, which suggested that the interactive relationship between IFN and NLRC5 might form a feedback loop to regulate the expression of MHC-I. More recently, Chen et al. utilized a specific pMHC-I-guided CRISPR-Cas9 screening method to identify crucial MHC-I regulators and found that a key inhibitory molecular SUSD6/TMEM127/WWP2 axis, in which SUSD6, TMEM127, and MHC-I forms a trimolecular complex to recruit WWP2 for MHC-I ubiquitination and lysosomal degradation. This process resulted in the decrease of MHC-I expression and impaired anticancer immunity, which subsequently shortens the cancer patients’ survival periods [15]. This newly-identified inhibitory pathway may be a novel therapeutic target for reinvigoration of CD8+ T cells.
Moreover, the expression of MHC-I is also regulated by epigenetic mechanisms, such as histone deacetylation, DNA methylation, and polycomb repressive complex 2 (PRC2)-mediated histone 3 lysine 27 trimethylation (H3K27me3), which help limit the expression of MHC-I. In consistency, administration of the inhibitors for these processes, including DNA Methyltransferases inhibitor (DNMTi) [16] and Histone methyltransferase [17, 18] could up-regulate the expression of MHC-I complex. Non-coding RNA (ncRNA) is the other important part of epigenetic modulators, which plays major roles in regulating the expression of MHC-I. Hu et al. identified that miRNA 34a (miR34a) is enriched in abundance in the develo** hippocampal neurons, which targets at the 3′UTR site of MHC-I mRNA, thus decreasing the expression of MHC-I and facilitating the normal development of neural morphology in develo** hippocampal neurons [19]. Notably, in the context of cancer, miRNAs also participate in the regulation of MHC-I expression, which subsequently influences the process of anti-cancer immunity. Zheng et al. found that miR-148-3p serves as an oncogene by targeting on calnexin pathway, which suppresses the surface expression of MHC-I on tumor cells and inhibits the CD8+ T cell-dominated anticancer immunity in colorectal cancer [36]. Recent studies have demonstrated that the tumor microenvironment alters the cytokines produced by NK cells, leading to changes in tumor progression. NK cells pre-exposed to tumor cells expressing MHC-I promote host extramedullary myelopoiesis, which is partly related to TNF-α secretion by tumor-experience NK cells [37].
Lastly, the ‘missing-self recognition’ by NK cell surveillance may be circumvented by tumors. Current NK-cell-based immune therapies have focused on the interaction between NK cells and tumor cells. Understanding the features of the tumor is essential to maximizing the NK cells’ untapped potential and develo** clinical therapeutic interventions.
Tumor-associated macrophages (TAMs)
TAMs have been identified as a key intertumoral regulator with pro- and anti-tumorigenic dual functions. TAMs are highly complex and plastic immune cells in the TME. It can be divided into M1 and M2 subtypes after activation. The M1 subtype is a classical anti-tumor killer cell, whereas the M2 subtype is an immunosuppressive subtype that expresses a range of suppressive cytokines. The structural molecules of the MHC-I complex, the heavy chain and B2M, can interact with TAMs to exert immunosuppressive effects.
A review summarized the roles of heavy chain for TAMs. The HLA-A, B, C in cancer cells suppress macrophage activation or stimulate alternative macrophage differentiation via the leukocyte lg-like receptors (LILRs) family. They also reported that the heavy chain in TAMs conducts the immunosuppressive function through inhibiting NK and T cell activities or releasing cytokines [38]. Similarly, the activation of macrophages can be adversely regulated by MHC-I light chain B2M. According to Li et al., B2M promotes the M2 phenotype in TAMs. Furthermore, they confirmed that M2 polarization is attributed to the B2M-induced TGF-β activation of the PI3K/ATK signaling in TAMs [39]. Recently, Barkal et al. elucidated that MHC-I can directly inhibit the phagocytic activity of TAMs. B2M of the MHC-I molecules can combine with the inhibitory receptor LILRB1 on the surface of macrophages, protecting cancer cells from phagocytosis [40].
These researches provided new insights into the function of MHC-I in the intrinsic immunity system. MHC-I molecules have the potential to reprogram TME into a tumor-promoting state. To disturb the macrophage-mediated tumor cell killing effect, MHC-I molecules can directly interact with immune cells or secrete immunosuppressive cytokines indirectly.
Other cells
Dendritic cells (DCs) are well-known antigen-presenting cells in the immune response. The theory of antigen cross-presentation, once it was put forward, contributed to a deeper comprehension of DCs in the TME. Cross-presentation is a critical process by which DCs mediate antigen presentation in non-DC infections and activate CD8+ T cells. DCs take up tumor cells that are perishing and go through a maturation process. The antigens are processed and loaded onto MHC-I and MHC-II for presentation to CD8+ T cells and CD4+ T cells, respectively, as they migrate to the lymph node [41]. It is disappointing to note that DCs in TME might exhibit cross-presentation abnormalities that can be attributed to oxidized lipid. It precluded cross-presentation by the interaction between ox-tr-LB and the chaperone HSP70, which led to the translocation of the MHC-I to endosomes rather than the cell surface [42]. In fact, further research is still required to determine the processes underlying immune tolerance or lymph node metastases linked to DC cells.
Myeloid-derived suppressor cells (MDSCs) are well-known as immunosuppressive participants in TME [7]. Yamamoto et al. revealed that MHC-I molecules will enter lysosomal degradation through the cargo receptor NBR1-mediated autophagy process in pancreatic ductal adenocarcinoma (PDAC) cells. It leads to reduced MHC-I expression on the cell surface and impaired immunotherapy [7]. Similarly, Fang et al. discovered that the interaction between MAL2 and MHC-I molecules and RAB proteins, that is, endosome-related proteins, converts to lysosomal degradation in breast cancer cells [76]. Thus, it inhibited tumor antigen presentation and decreased CD8+ T cell infiltration. They also observed that MAL2 deletion can significantly enhance the infiltration of CD8+ T cells in the preclinical model. Consistently, suppression of PGRN restores MHC-I expression in PDAC cells by decreasing lysosomal activity and autophagosome degradation [77]. Notably, the authors constructed a PDAC mouse model using a model antigen, LCMV-GP33. Tumors with LCMV-GP33 are sensitized to gp33-TCR transgenic T cells, regaining tumor immunogenicity and tumor antigen-specific cytotoxicity. YTHDF1 deficiency constrains lysosomal-related proteolysis of MHC-I molecules. In order to target YTHDF1 in vivo, Lin et al. developed a system for exosome-mediated CRISPR/Cas9 delivery, which leads to YTHDF1 depletion and restores tumor immune surveillance [78]. These studies persuaded us that decreasing CD8+ T cell infiltration and immunosuppression may occur by exploiting the autophagy pathway. Additionally, we can speculate that targeting this process can restore tumor immune surveillance and make patients more sensitive to ICB therapy.
Immune cells
Classical MHC-I-related immunoregulatory mechanisms include the genetic, transcriptional, translational, and post-translational levels. A series of findings suggested that cancer cells will directly suppress the expression of MHC-I molecules or interfere with the molecules involved in the antigen processing and presentation pathway. It impairs the CD8+ T cell-mediated immune response and flees from immune surveillance. In addition to directly or indirectly affecting T cells, the suppressive effect of MHC-I on the intrinsic immune system should not be underestimated.
MHC-I can exert its immunosuppressive function through a variety of immune cells. For example, this can be achieved by engaging with the inhibitory receptor KIR on NK cells or by targeting macrophages through various inhibitory pathways. In addition, the inhibitory cells Tregs and MDSCs suppress the cytotoxicity of NK cells and T cells, respectively. In the previous section, we offered a more extensive review.
The association between MHC-I molecules and pathological characteristics of human cancers
The dual function of MHC-I molecules in tumor immunity leads to different immune states in patients, which will affect the growth and metastasis of tumor cells and eventually influence the pathological characteristics of human cancers as well as the prognosis of patients. Therefore, MHC-I molecules might be utilized to establish the molecular stage of cancer, and have great application potential as biomarkers or models to predict tumor diagnosis, prognosis, recurrence, and immunotherapy response.
Traditionally, it is believed that cancer cells with downregulated or deficient surface MHC-I molecules are more likely to escape T cell surveillance, thus leading to a poor prognosis. Studies revealed classical MHC-I molecules and B2M may be an immunological prognostic factor for varied cancers [79,80,81,82,83]. As for immunotherapy, downregulation of classical MHC-I has been identified as a risk factor for recurrence in bladder cancer patients treated with BCG immunotherapy [84]. Zhao et al. revealed that a high level of B2M expression could enhance the anticancer immune response [85]. Other studies reported the opposite results due to the dual function of MHC-I molecules. They considered that cancer cells lacking MHC-I molecules were more sensitive to NK cells. This may be the reason for the good prognosis of low-level expression of HLA [86, 87]. Intriguingly, Watson demonstrated that the high expression or absence of HLA correlates with a better prognosis than the low expression of HLA in colorectal cancer [88]. In summary, CD8+ T cells can attack cancer cells with high HLA expression, whereas TAMs and NK cells can eliminate cancer cells without MHC-I expression. Low expression of MHC-I might escape both acquired and inherent immunity. We speculate that the reason for the opposite prognostic results may be relative expression. Another possibility is that the heterogeneity also causes different tumor microenvironment, ultimately leading to different prognostic outcomes for patients.
As for non-classical MHC-I, it is usually considered to be upregulated in cancer. Physiologically, a reasonable explanation is that nonclassical MHC-I molecules play an immune tolerance role to protect embryos from maternal immune system attack [89], cancers may exploit this property for immune escape. HLA-E [90,91,92], HLA-F [93,94,95] and HLA-G [96,97,98], which have been identified as novel immune checkpoints, can bind to the inhibitory receptor of immune cells to induce immune tolerance and are regarded as prognostic factors. Morinaga et al. reported that the combination of HLA-E expression and NK status can be a more sensitive prognostic biomarker in advanced gastric cancer [90]. HLA-G is a significant prognostic indicator of colorectal cancer, and the possible mechanism is the binding of HLA-G to its suppressive immune checkpoints ILT-2 and ILT-4 [96, 98, 99]. Wu et al. have revealed that high HLA-F expression is closely associated with local recurrence and distant metastasis of nasopharyngeal carcinoma [93]. A study reported that HLA-G is a biomarker of tumor cell susceptibility to therapeutic agents by the immune response or treatment [97].
With the rapid development of bioinformatics, numerous hub genes and prognostic models were identified based on the MHC-I-related genes [82, 100], it showed great significance for prognostic prediction and guiding immunotherapy. These results inspired us to believe that the impact of MHC-I molecules on pathological characteristics varies considerably by cancer. Deeper mechanisms and larger cohorts should be enrolled to determine the pathological characteristics of MHC-I molecules in different cancers.
Therapeutic strategies and advances targeted on MHC-I
Immunotherapy
Immune checkpoint blockade
ICB therapy is regarded as a milestone in cancer immunotherapy, particularly the anti-CTLA4, anti-PD-1, and anti-PD-L1 drugs. Though ICB therapy has maintained tumor shrinkage and prolonged life expectancy in some patients, non-response and resistance remain barriers to treatment. Numerous mechanisms have been investigated, with the antigen presentation defect being a key one.
Recently, HLA-E:CD94/NKG2A has been identified as a novel immune checkpoint. Targeting this immune checkpoint can induce the anti-tumor activity of CD8+ T cells and NK cells [27, 105]. The most promising anti-NKG2A mAb, called monalizumab, prevents the interaction between NKG2A and HLA-E and exhibits a potent therapeutic impact on several cancer types, such as NSCLC, CRC, and SCCHN [106]. Disappointingly, the highly anticipated monalizumab for head and neck cancer finally failed in the Phase III clinical trial in August 2022. Its clinical therapeutic effect in other cancers still requires further clinical trials to validate.
Alteration of genes that encoding MHC-I molecules will affect ICB efficacy. Previous research documented LOH at HLA in resistant tumor that are treated with T cell transfer therapy [107]. Later, relevant clinical research has reported that HLA homozygosity and LOH in cancer patients represent a genetic barrier to effective ICB therapy [108]. A similar outcome could be demonstrated in B2M, namely that B2M LOH is prevalent in cancer patients who have a poor response to or non-response to ICB therapy [50, 109]. In addition, genetic mutations in B2M, resulting in the formation of defective HLA, may also be associated with acquired resistance to ICB treatment [110, 111].
Impaired antigen presentation may be a mechanism of resistance to ICB therapy, including HLA heavy chain, light chain B2M, and other antigen presentation components [110, 112, 113]. This is associated with increased presentation of tumor antigen as well as T cell recognition. Therefore, upregulation of MHC-I molecules expression is a promising therapeutic strategy to enhance the synergy with ICB. PCSK9 inhibition potentiates MHC-I expression, which promotes T cells to infiltrate the tissue, making the tumor more sensitive to the immune checkpoints [114]. In order to conduct this test, Liu et al. used two anti-PCSK9 antibodies (evolocumab and alirocumab), which have been licensed for treating hypercholesterolemia. The combination of ICB and anti-PCSK9 antibody has to be tested in clinical use. Dysregulation of signaling pathways, such as interferon (IFN) pathway [113, 115,116,117], MAPK pathway [69], STAT pathway [117,118,119] and EGFR pathway [120], may result in aberrant MHC-I molecules production. In this condition, different molecules in the signaling pathway have been the targets of drugs. IFN-γ could induce the HLA expression [117]. Kang reported that the MEK inhibitor trametinib could block the MAPK pathway and thereby increase the MHC-I, which may lead to improved ICB efficacy [121]. In addition, bortezomib, a proteasome inhibitor (targeted at STAT1), can enhance the expression of HLA [118]. Lenvatinib restored STAT1 phosphorylation and the expression of B2M. When paired with an anti-PD-1 antibody, lenvatinib had stronger anticancer activity [119].
The expression of MHC-I molecules can be regulated epigenetically, typically by the use of epigenetic inhibitors, such as DNMTi [55, 56], HDACi [55, 62, 63] and EZH2 inhibitor [58]. Notably, Ugurel et al. reported 4 cases with non-response to ICB therapy enhanced HLA expression with the combination with HDACi [64]. Combining EZH2 inhibitior with ICB reduces tumor growth by changing the expression of B2M [58]. Therefore, we can conclude that these targeted drugs and epigenetic drugs restore MHC-I molecules expression and thus enhance their synergistic effect with ICB. It urged that further research be conducted to confirm the specific mechanisms.
Additionally, the effectiveness of ICB therapy will be enhanced by using it in combination with chemotherapy, radiotherapy, and other strategies due to the upregulation of HLA.
Abnormal expression of immune checkpoint molecules in human cancers has become the dominant mechanism by which cancer cells escape the immune response and ultimately lead to tumor development. With in-depth and extensive research, MHC-I molecules have been identified as novel immune checkpoint molecules [97]. There is an urgent need to explore the role of MHC-I molecules as immune checkpoints in ICB therapies. Perhaps medications that specifically target these immune checkpoints will become available in the future.
Cancer vaccines
Cancer vaccines, which can be classified as prophylactic and therapeutic vaccines, offer a promising way to improve the anti-tumor response. Since Alvaro Morales used Bacillus Calmette Guerin (BCG) to treat superficial bladder cancer in 1976, therapeutic vaccines have emerged [122]. Nowadays, BCG is still the standard treatment for non-muscle invasive bladder cancer. Besides, some research uncovered that HLA status plays a crucial role in the clinical characteristics of bladder cancer patients following BCG therapy [123,124,125].
Recently, some scientists conducted studies on personalized vaccines. This ushers in a new age of cancer vaccines and brings new research directions worth exploring. Targeting tumor neoantigens with high affinity for HLA has become acknowledged as feasible targets for personalized therapeutic vaccines. Tumor neoantigen vaccination can enhance the anti-tumor immune response, notably neoantigen-specific T cells [126,127,128]. Therapeutic vaccines have improved specificity and safety compared to conventional radiation and chemotherapy therapies.
In fact, the MHC-I genotype of patients has been shown to affect the clinical outcome [129,130,131]. Frameshift mutations in calreticulin occur in myeloproliferative neoplasm. However, MHC-I alleles that exhibit mutant neoepitopes with high affinity are underrepresented. They utilized a modified heteroclitic peptide vaccination method for patients to efficiently elicit T cell response [132]. Moreover, one of the possible causes of cancer vaccine resistance is abnormal MHC-I molecule expression. Cancer vaccination with ODN1862 adjuvant was only marginally effective in MHC-I-negative models that deleted the B2M gene in TC-1 cell line [133]. Cao et al. have developed a nanovaccine named HA-OVA-AuNPs that enhances proteasome activity and downstream MHC-I antigen presentation [134].
Neoantigen-specific T cells are limited in TME, expanding cytotoxic T lymphocytes (CTLs) against tumor cells may become a prospective strategy. Montfoort et al. revealed that therapeutic cancer vaccines targeting HLA-E:NKG2A might be a potential strategy to activate CD8+ T cell and NK cell immunity in microenvironment [29]. Intriguingly, some therapeutic cancer vaccines aim to disguise tumor cells as human CMV-infected cells by showing CMV-pMHC on their surface, allowing CMV-specific CTL to detect and lyse them [135, 136]. Through these strategies, bystander T cells [137] that recognize antigens unrelated to cancer were fully mobilized to attack tumor cells.
In addition, cancer vaccines combined with other therapy strategies aim to foster a favorable immune environment and enhance immune responses. Zhu et al. created the first engineered Lactococcus lactis-based cancer vaccine using probiotic loading neoantigens for ongoing activation [138]. Combining personalized cancer vaccines with PD-1, PD-L1, and CTLA4 inhibitors is now being used in different clinical trials to treat various cancer types [131]. An ICB immunotherapy, atezolizumab, and an individualized neoantigen vaccine with up to 20 MHC-I and MHC-II restricted neoantigens in nanoparticles autogene cevumeran are being tested in a phase I clinical trial [139].
Chimeric antigen receptor T cell (CAR-T) therapy
CAR-T therapy mediates non-MHC-restrictive cancer cell death by transducing CAR on the surface of T cells to detect and eliminate cancer cells.
Utilizing allogeneic CAR-T cells can be an efficient strategy to overcome the poor quality and quantity of autologous T cells. However, when HLA divergence occurs, the host immune system rejects allogeneic T cells. This obstacle can be solved by the CRISPR/Cas9 system to create negative HLA T cells [140]. Notably, negative MHC-I might activate NK cells, which then act as the main cause of CAR-T cell death [141]. Therefore, more studies are required to find out how to prevent them from killing CAR-T cells.
There are still studies being done on how to improve the efficiency of CAR-T cells. It has been demonstrated that INF-γ can be secreted by CAR-T cells, which will upregulate MHC molecules in cancer cells [142]. Moreover, combination therapy is a way to enhance the effect of CAR-T therapy. Radiation therapy can increase MHC-I expression, enabling T cells to more readily detect cancer cells. Therefore, combining CAR-T therapy and radiation therapy may have a synergistic impact [143, 144]. These findings provide an essential guide for the synergistic benefit of combining CAR-T therapy with conventional anticancer therapies targeted at MHC-I molecules.
Cytokine therapy
Cytokines have powerfully modulated functions in cell communication, affecting the tumor immune microenvironment and triggering anticancer responses. The majority of researchers believe that MHC-I-deficient cancer cells are killed by NK cells because they cannot bind inhibitory receptors on NK cells [145]. In fact, the cancer cells can bypass immune surveillance by NK cells, but cytokine therapy with IL-12 and IL-18, or H9 can reverse the anergic NK cells [36]. IFN molecules are promising cytokines targeted at MHC-I because they prompt DCs or T cells to eliminate cancer cells through upregulating MHC-I molecules [146]. Combination therapy with ICB has been done in clinical trials [147].
Radiotherapy
Radiotherapy provides an effective therapeutic strategy to eliminate cancer cells. Biological mechanisms have been proven to include DNA damage response, immune modulation, and altering TME in cancer cells [148]. Notably, MHC-I could be upregulated by radiation, which makes it possible to improve antigen presentation and increase the recognition capacity and cytotoxicity of T cells [149,150,151,152].
Some researchers tried to demonstrate the complicated mechanism. Radiation activated IFN signaling, which enhanced the expression of MHC-I and antigen presentation, overcoming the resistance to ICB therapy [115]. In addition, radiation might induce the expression of NLRC5, impacting MHC-I expression and easing ICB limitation [151]. DNA damage has been proven to cause elevated MHC-I [153]. Whether the DNA damage response caused by radiotherapy still deserves further study and exploration.
Combination therapy of radiotherapy with other therapies can be an emerging strategy for treating cancer. Firstly, combined with adoptive T cell therapy, radiotherapy boosts the efficacy of the anticancer response by upregulating MHC-I expression and enhancing the sensitivity to cytotoxicity T cells [152]. Similarly, radiotherapy combined with GM-CSF vaccination is more effective in gliomas [154]. We can hypothesize that increasing MHC-I expression by radiotherapy may be the mechanism for synergistic effect with other immunotherapies but the underlying mechanisms require experiments to verify. On the other hand, radiotherapy can induce more neoantigen release and prompt T cell infiltration [128], which can circumvent the problem of insufficient neoantigen presentation and failure to activate the T cells. It indicates that radiotherapy can be combined with other therapies that can upregulate MHC-I. From this perspective, radiotherapy may be unsuitable for MHC-I-deficient cancers.
Conventional chemotherapeutics and targeted agents
There is little research on how MHC-I is impacted by conventional chemotherapeutics and targeted agents. Enhancing MHC-I expression is the most common mechanism to promote anticancer responses in these therapies. Cisplatin, a DNA damaging agent, is widely used in clinical anticancer practice. Accumulating evidence points out that it has immunomodulatory effects and elevates MHC-I expression in different types of cancers [155], possibly related to the activation of IFN-β, NF-κB signaling pathway [116]. Moreover, cisplatin also upregulates MHC-I in antigen-presenting cells, such as DC cells [156]. Zhou et al. found that the lysine acetyl transferases CBP regulate MHC-I expression and neoantigen amounts in human cancer. DNA damaging drugs upregulate MHC-I dependent on activation of NF-κB [153]. Liu et al. reported that cyclophosphamide, oxaliplatin, and gemcitabine stimulate the expression of HLA by cancer cells [157]. Similar results were also found in gefitinib [158], MAPK inhibitors [159] and 5-Fluorouracil [160]. The study by Liu et al. similarly showed that PCSK9 Binds to pMHC-I to promote its degradation in lysosomes [114]. A solid foundation has been established for the introduction of PCSK9 inhibitors, a lipid-lowering medication with clinical approval, into oncology immunotherapy. They also show the evidences on targeting PCSK9 also improves the anti-tumor activity of PD-1 immune checkpoint. A study intriguingly suggests that downregulated MHC-I expression caused by imatinib is another way to boost immunity attribute to the relocation of NK cells to cancer [161]. In addition to altering the immunomodulatory effect by affecting MHC-I, chemotherapy can directly activate CD8+ T cells in an MHC-I-independent manner [162].
Conclusions
In conclusion, MHC-I and its related molecules are crucial participators in the tumor microenvironment. Classical antigen presentation functions by MHC-I maintain the basic anticancer immunity of CD8+ T cells, which endows MHC-I with an indispensable place in the battle against tumor cells. Nevertheless, emerging studies generally uncovered the immune escape capabilities dominated by MHC-I in cancer. Multiple factors coherently modulate the expression and function of MHC-I in the context of cancer. MHC-I-related pathways may be a promising and efficacious target for synergizing with cancer immunotherapy. However, the identification and explanation of crucial molecules modulating the MHC-I functioning directions and their definite mechanisms are warranted. We hope this review would help summarize the latest and key knowledge of MHC-I in cancer, and, more importantly, inspire more studies on the therapeutic exploration based on MHC-I molecules.
Data Availability
Not applicable.
References
Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. 2022;22:751–64.
Wong-Benito V, de Rijke J, Dixon B. Antigen presentation in vertebrates: structural and functional aspects. Dev Comp Immunol. 2023;144:104702.
Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, Moeller A, Tampe R. Structure of the human MHC-I peptide-loading complex. Nature. 2017;551:525–8.
Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37:724–37.
van Hateren A, Elliott T. The role of MHC I protein dynamics in tapasin and TAPBPR-assisted immunopeptidome editing. Curr Opin Immunol. 2021;70:138–43.
Sykulev Y. Factors contributing to the potency of CD8(+) T cells. Trends Immunol 2023.
Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. Autophagy promotes immune evasion of Pancreatic cancer by degrading MHC-I. Nature. 2020;581:100–5.
Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion through loss of MHC Class I Antigen Presentation. Front Immunol. 2021;12:636568.
Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet. 2010;11:47–59.
Creech AL, Ting YS, Goulding SP, Sauld JFK, Barthelme D, Rooney MS, Addona TA, Abelin JG. The role of Mass Spectrometry and Proteogenomics in the Advancement of HLA Epitope Prediction. Proteomics. 2018;18:e1700259.
Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–81.
Benko S, Kovacs EG, Hezel F, Kufer TA. NLRC5 functions beyond MHC I regulation-what do we know so far? Front Immunol. 2017;8:150.
Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Buning H, Schwarzenbacher R, Kufer TA. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285:26223–32.
Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. Opposing functions of Interferon Coordinate Adaptive and Innate Immune responses to Cancer Immune Checkpoint Blockade. Cell. 2019;178:933–948e914.
Chen X, Lu Q, Zhou H, Liu J, Nadorp B, Lasry A, Sun Z, Lai B, Rona G, Zhang J et al. A membrane-associated MHC-I inhibitory axis for cancer immune evasion. Cell 2023.
Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, Zahnow CA, Nickels ML, Liu F, Tantawy MN, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in Breast cancer. Nat Commun. 2018;9:248.
Ren J, Li N, Pei S, Lian Y, Li L, Peng Y, Liu Q, Guo J, Wang X, Han Y et al. Histone methyltransferase WHSC1 loss dampens MHC-I antigen presentation pathway to impair IFN-gamma-stimulated antitumor immunity. J Clin Invest 2022, 132.
Sun T, Li Y, Yang W, Wu H, Li X, Huang Y, Zhou Y, Du Z. Histone deacetylase inhibition up-regulates MHC class I to facilitate cytotoxic T lymphocyte-mediated Tumor cell killing in glioma cells. J Cancer. 2019;10:5638–45.
Hu Y, Pei W, Hu Y, Li P, Sun C, Du J, Zhang Y, Miao F, Zhang A, Shen Y, Zhang J. MiR34a regulates neuronal MHC class I molecules and promotes primary hippocampal neuron dendritic growth and branching. Front Cell Neurosci. 2020;14:573208.
Zheng J, Yang T, Gao S, Cheng M, Shao Y, ** Y, Guo L, Zhang D, Gao W, Zhang G, et al. miR-148a-3p silences the CANX/MHC-I pathway and impairs CD8(+) T cell-mediated immune Attack in Colorectal cancer. FASEB J. 2021;35:e21776.
Mari L, Hoefnagel SJM, Zito D, van de Meent M, van Endert P, Calpe S, Sancho Serra MDC, Heemskerk MHM, van Laarhoven HWM, Hulshof M, et al. microRNA 125a regulates MHC-I expression on Esophageal Adenocarcinoma Cells, Associated with suppression of Antitumor Immune response and poor outcomes of patients. Gastroenterology. 2018;155:784–98.
Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298–312.
Dersh D, Holly J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol. 2021;21:116–28.
Jongsma MLM, de Waard AA, Raaben M, Zhang T, Cabukusta B, Platzer R, Blomen VA, Xagara A, Verkerk T, Bliss S, et al. The SPPL3-Defined glycosphingolipid repertoire orchestrates HLA class I-Mediated Immune responses. Immunity. 2021;54:132–150e139.
Ito Y, Pan D, Zhang W, Zhang X, Juan TY, Pyrdol JW, Kyrysyuk O, Doench JG, Liu XS, Wucherpfennig KW. Addressing Tumor heterogeneity by sensitizing resistant Cancer cells to T cell-secreted cytokines. Cancer Discov. 2023;13:1186–209.
Westrich JA, Vermeer DW, Silva A, Bonney S, Berger JN, Cicchini L, Greer RO, Song JI, Raben D, Slansky JE, et al. CXCL14 suppresses human papillomavirus-associated Head and Neck cancer through antigen-specific CD8(+) T-cell responses by upregulating MHC-I expression. Oncogene. 2019;38:7166–80.
Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Blery M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK Cells. Cell. 2018;175:1731–1743e1713.
Abd Hamid M, Wang RZ, Yao X, Fan P, Li X, Chang XM, Feng Y, Jones S, Maldonado-Perez D, Waugh C, et al. Enriched HLA-E and CD94/NKG2A Interaction limits Antitumor CD8(+) tumor-infiltrating T lymphocyte responses. Cancer Immunol Res. 2019;7:1293–306.
van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, Andre P, et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer vaccines. Cell. 2018;175:1744–1755e1715.
Lerner EC, Woroniecka KI, D’Anniballe VM, Wilkinson DS, Mohan AA, Lorrey SJ, Waibl-Polania J, Wachsmuth LP, Miggelbrink AM, Jackson JD et al. CD8(+) T cells maintain killing of MHC-I-negative Tumor cells through the NKG2D-NKG2DL axis. Nat Cancer 2023.
Eugene J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, Leveque E, Meurette G, Podevin J, Matysiak T, et al. The inhibitory receptor CD94/NKG2A on CD8(+) tumor-infiltrating lymphocytes in Colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/beta2m overexpression. Mod Pathol. 2020;33:468–82.
Liu X, Song J, Zhang H, Liu X, Zuo F, Zhao Y, Zhao Y, Yin X, Guo X, Wu X, et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating Tumor cells from NK cell surveillance. Cancer Cell. 2023;41:272–287e279.
Krijgsman D, Roelands J, Hendrickx W, Bedognetti D, Kuppen PJK. HLA-G: a New Immune Checkpoint in Cancer? Int J Mol Sci 2020, 21.
Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol. 2023;23:90–105.
Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, Tsoucas D, Franz B, May KF Jr., Harvey CJ, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven Tumor immunity. Science. 2018;359:1537–42.
Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, Deng W, Ring AM, Fischer S, Garcia KC, Raulet DH. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest. 2014;124:4781–94.
Neo SY, **g X, Tong L, Tong D, Gao J, Chen Z, De Los Santos MC, Burduli N, De Souza Ferreira S, Wagner AK et al. Tumor MHC class I expression alters cancer-associated myelopoiesis driven by host NK cells. J Immunother Cancer 2022, 10.
Marchesi M, Andersson E, Villabona L, Seliger B, Lundqvist A, Kiessling R, Masucci GV. HLA-dependent tumour development: a role for tumour associate macrophages? J Transl Med. 2013;11:247.
Li D, Zhang Q, Li L, Chen K, Yang J, Dixit D, Gimple RC, Ci S, Lu C, Hu L, et al. beta2-Microglobulin maintains Glioblastoma Stem cells and induces M2-like polarization of Tumor-Associated macrophages. Cancer Res. 2022;82:3321–34.
Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, George BM, Markovic M, Ring NG, Tsai JM, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19:76–84.
Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, et al. Human CD141+ (BDCA-3) + dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–60.
Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, Hashimoto A, Kapralov A, Amoscato A, Angelini R, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017;8:2122.
Li T, Liu T, Zhu W, **e S, Zhao Z, Feng B, Guo H, Yang R. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: current progress and new prospects. Clin Med Insights Oncol. 2021;15:11795549211035540.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8 + T cell tolerance in cancer. Nat Med. 2007;13:828–35.
Koopman LA, Corver WE, van der Slik AR, Giphart MJ, Fleuren GJ. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in Cervical cancer. J Exp Med. 2000;191:961–76.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
Wang H, Liu B, Wei J. Beta2-microglobulin(B2M) in cancer immunotherapies: biological function, resistance and remedy. Cancer Lett. 2021;517:96–104.
Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J Immunother Cancer 2021, 9.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. Allele-specific HLA loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171:1259–1271e1211.
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136.
Ye Q, Shen Y, Wang X, Yang J, Miao F, Shen C, Zhang J. Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens. 2010;75:30–9.
Qifeng S, Bo C, **ngtao J, Chuanliang P, **aogang Z. Methylation of the promoter of human leukocyte antigen class I in human esophageal squamous cell carcinoma and its histopathological characteristics. J Thorac Cardiovasc Surg. 2011;141:808–14.
Hasim A, Abudula M, Aimiduo R, Ma JQ, Jiao Z, Akula G, Wang T, Abudula A. Post-transcriptional and epigenetic regulation of antigen processing machinery (APM) components and HLA-I in cervical cancers from Uighur women. PLoS ONE. 2012;7:e44952.
Snahnicanova Z, Kasubova I, Kalman M, Grendar M, Mikolajcik P, Gabonova E, Laca L, Caprnda M, Rodrigo L, Ciccocioppo R, et al. Genetic and epigenetic analysis of the beta-2-microglobulin gene in microsatellite instable Colorectal cancer. Clin Exp Med. 2020;20:87–95.
Rodems TS, Heninger E, Stahlfeld CN, Gilsdorf CS, Carlson KN, Kircher MR, Singh A, Krueger TEG, Beebe DJ, Jarrard DF, et al. Reversible epigenetic alterations regulate class I HLA loss in Prostate cancer. Commun Biol. 2022;5:897.
Moufarrij S, Srivastava A, Gomez S, Hadley M, Palmer E, Austin PT, Chisholm S, Diab N, Roche K, Yu A, et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases Tumor burden in Ovarian cancer. Sci Rep. 2020;10:3470.
Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, Azidis-Yates E, Vassiliadis D, Bell CC, Gilan O, et al. An evolutionarily conserved function of polycomb silences the MHC Class I Antigen Presentation Pathway and enables Immune Evasion in Cancer. Cancer Cell. 2019;36:385–401e388.
Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 enhances Antigen Presentation, Antitumor Immunity, and circumvents Anti-PD-1 resistance in Head and Neck Cancer. Clin Cancer Res. 2020;26:290–300.
Li T, Du D, Zhang D, Lin Y, Ma J, Zhou M, Meng W, ** Z, Chen Z, Yuan H, et al. CRISPR-based targeted haplotype-resolved assembly of a megabase region. Nat Commun. 2023;14:22.
Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, tapasin genes and MHC class I antigen presentation by Melanoma cells. Cancer Immunol Immunother. 2008;57:647–54.
Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, Woods DM, Barrios K, Powers J, Sahakian E, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015;9:1447–57.
Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016;7:7390–402.
Souri Z, Jochemsen AG, Versluis M, Wierenga APA, Nemati F, van der Velden PA, Kroes WGM, Verdijk RM, Luyten GPM, Jager MJ. HDAC inhibition increases HLA Class I expression in Uveal Melanoma. Cancers (Basel) 2020, 12.
Ugurel S, Spassova I, Wohlfarth J, Drusio C, Cherouny A, Melior A, Sucker A, Zimmer L, Ritter C, Schadendorf D, Becker JC. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer Immunol Immunother. 2019;68:983–90.
Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28:239–60.
Gao J, Shi LZ, Zhao H, Chen J, **ong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma pathway genes in Tumor cells as a mechanism of resistance to Anti-CTLA-4 therapy. Cell. 2016;167:397–404e399.
Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, Vega-Crespo A, Parisi G, Champekar A, Nguyen C et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in Melanoma. Sci Transl Med 2020, 12.
Marijt KA, Sluijter M, Blijleven L, Tolmeijer SH, Scheeren FA, van der Burg SH, van Hall T. Metabolic stress in cancer cells induces immune Escape through a PI3K-dependent blockade of IFNgamma receptor signaling. J Immunother Cancer. 2019;7:152.
Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, Mondello P, Han JE, Jarvis CA, Ulmert D, et al. Kinase regulation of human MHC class I molecule expression on Cancer cells. Cancer Immunol Res. 2016;4:936–47.
Concha-Benavente F, Srivastava RM, Ferrone S, Ferris RL. EGFR-mediated Tumor immunoescape: the imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology. 2013;2:e27215.
Sivaram N, McLaughlin PA, Han HV, Petrenko O, Jiang YP, Ballou LM, Pham K, Liu C, van der Velden AW, Lin RZ. Tumor-intrinsic PIK3CA represses Tumor immunogenecity in a model of Pancreatic cancer. J Clin Invest. 2019;129:3264–76.
Chandrasekaran S, Sasaki M, Scharer CD, Kissick HT, Patterson DG, Magliocca KR, Seykora JT, Sapkota B, Gutman DA, Cooper LA, et al. Phosphoinositide 3-Kinase signaling can modulate MHC class I and II expression. Mol Cancer Res. 2019;17:2395–409.
Yang W, Li Y, Gao R, **u Z, Sun T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/beta-catenin signaling pathway. Oncogene. 2020;39:1098–111.
Huang L, Malu S, McKenzie JA, Andrews MC, Talukder AH, Tieu T, Karpinets T, Haymaker C, Forget MA, Williams LJ, et al. The RNA-binding protein MEX3B mediates resistance to Cancer Immunotherapy by Downregulating HLA-A expression. Clin Cancer Res. 2018;24:3366–76.
Colangelo T, Polcaro G, Ziccardi P, Pucci B, Muccillo L, Galgani M, Fucci A, Milone MR, Budillon A, Santopaolo M, et al. Proteomic screening identifies calreticulin as a miR-27a direct target repressing MHC class I cell surface exposure in Colorectal cancer. Cell Death Dis. 2016;7:e2120.
Fang Y, Wang L, Wan C, Sun Y, Van der Jeught K, Zhou Z, Dong T, So KM, Yu T, Li Y et al. MAL2 drives immune evasion in Breast cancer by suppressing Tumor antigen presentation. J Clin Invest. 2021;131.
Cheung PF, Yang J, Fang R, Borgers A, Krengel K, Stoffel A, Althoff K, Yip CW, Siu EHL, Ng LWC, et al. Progranulin mediates immune evasion of pancreatic ductal adenocarcinoma through regulation of MHCI expression. Nat Commun. 2022;13:156.
Lin W, Chen L, Zhang H, Qiu X, Huang Q, Wan F, Le Z, Geng S, Zhang A, Qiu S, et al. Tumor-intrinsic YTHDF1 drives immune evasion and resistance to immune checkpoint inhibitors via promoting MHC-I degradation. Nat Commun. 2023;14:265.
Homma I, Kitamura H, Torigoe T, Tanaka T, Sato E, Hirohashi Y, Masumori N, Sato N, Tsukamoto T. Human leukocyte antigen class I down-regulation in muscle-invasive Bladder cancer: its association with clinical characteristics and survival after cystectomy. Cancer Sci. 2009;100:2331–4.
Kitamura H, Honma I, Torigoe T, Asanuma H, Sato N, Tsukamoto T. Down-regulation of HLA class I antigen is an Independent prognostic factor for clear cell renal cell carcinoma. J Urol. 2007;177:1269–72. discussion 1272.
Iwayama Y, Tsuruma T, Mizuguchi T, Furuhata T, Toyota N, Matsumura M, Torigoe T, Sato N, Hirata K. Prognostic value of HLA class I expression in patients with Colorectal cancer. World J Surg Oncol. 2015;13:36.
Chen X, Xu R, He D, Zhang Y, Chen H, Zhu Y, Cheng Y, Liu R, Zhu R, Gong L, et al. CD8(+) T effector and immune checkpoint signatures predict prognosis and responsiveness to immunotherapy in Bladder cancer. Oncogene. 2021;40:6223–34.
Benevolo M, Mottolese M, Tremante E, Rollo F, Diodoro MG, Ercolani C, Sperduti I, Lo Monaco E, Cosimelli M, Giacomini P. High expression of HLA-E in colorectal carcinoma is associated with a favorable prognosis. J Transl Med. 2011;9:184.
Kitamura H, Torigoe T, Honma I, Sato E, Asanuma H, Hirohashi Y, Sato N, Tsukamoto T. Effect of human leukocyte antigen class I expression of Tumor cells on outcome of intravesical instillation of bacillus calmette-guerin immunotherapy for Bladder cancer. Clin Cancer Res. 2006;12:4641–4.
Zhao Y, Cao Y, Chen Y, Wu L, Hang H, Jiang C, Zhou X. B2M gene expression shapes the immune landscape of lung adenocarcinoma and determines the response to immunotherapy. Immunology. 2021;164:507–23.
Madjd Z, Spendlove I, Pinder SE, Ellis IO, Durrant LG. Total loss of MHC class I is an Independent indicator of good prognosis in Breast cancer. Int J Cancer. 2005;117:248–55.
Zhang H, Cui B, Zhou Y, Wang X, Wu W, Wang Z, Dai Z, Cheng Q, Yang K. B2M overexpression correlates with malignancy and immune signatures in human gliomas. Sci Rep. 2021;11:5045.
Watson NF, Ramage JM, Madjd Z, Spendlove I, Ellis IO, Scholefield JH, Durrant LG. Immunosurveillance is active in Colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer. 2006;118:6–10.
Costanzo V, Bardelli A, Siena S, Abrignani S. Exploring the links between cancer and placenta development. Open Biol 2018, 8.
Morinaga T, Iwatsuki M, Yamashita K, Matsumoto C, Harada K, Kurashige J, Iwagami S, Baba Y, Yoshida N, Komohara Y, Baba H. Evaluation of HLA-E expression combined with natural killer cell status as a prognostic factor for Advanced Gastric Cancer. Ann Surg Oncol. 2022;29:4951–60.
Zeestraten EC, Reimers MS, Saadatmand S, Goossens-Beumer IJ, Dekker JW, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon Cancer patients. Br J Cancer. 2014;110:459–68.
Hiraoka N, Ino Y, Hori S, Yamazaki-Itoh R, Naito C, Shimasaki M, Esaki M, Nara S, Kishi Y, Shimada K, et al. Expression of classical human leukocyte antigen class I antigens, HLA-E and HLA-G, is adversely prognostic in Pancreatic cancer patients. Cancer Sci. 2020;111:3057–70.
Wu B, Yang H, Ying S, Lu H, Wang W, Lv J, **ong H, Hu W. High HLA-F Expression Is a Poor Prognosis Factor in Patients with Nasopharyngeal Carcinoma. Anal Cell Pathol (Amst) 2018, 2018:7691704.
Lin A, Zhang X, Ruan YY, Wang Q, Zhou WJ, Yan WH. HLA-F expression is a prognostic factor in patients with non-small-cell Lung cancer. Lung Cancer. 2011;74:504–9.
Harada A, Ishigami S, Kijima Y, Nakajo A, Arigami T, Kurahara H, Kita Y, Yoshinaka H, Natsugoe S. Clinical implication of human leukocyte antigen (HLA)-F expression in Breast cancer. Pathol Int. 2015;65:569–74.
Chen QY, Chen YX, Han QY, Zhang JG, Zhou WJ, Zhang X, Ye YH, Yan WH, Lin A. Prognostic significance of Immune checkpoints HLA-G/ILT-2/4 and PD-L1 in Colorectal Cancer. Front Immunol. 2021;12:679090.
Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: an Immune Checkpoint Molecule. Adv Immunol. 2015;127:33–144.
Guo ZY, Lv YG, Wang L, Shi SJ, Yang F, Zheng GX, Wen WH, Yang AG. Predictive value of HLA-G and HLA-E in the prognosis of Colorectal cancer patients. Cell Immunol. 2015;293:10–6.
Ye SR, Yang H, Li K, Dong DD, Lin XM, Yie SM. Human leukocyte antigen G expression: as a significant prognostic indicator for patients with Colorectal cancer. Mod Pathol. 2007;20:375–83.
Wang KW, Wang MD, Li ZX, Hu BS, Wu JJ, Yuan ZD, Wu XL, Yuan QF, Yuan FL. An antigen processing and presentation signature for prognostic evaluation and immunotherapy selection in advanced gastric cancer. Front Immunol. 2022;13:992060.
Michelakos T, Kontos F, Kurokawa T, Cai L, Sadagopan A, Krijgsman D, Weichert W, Durrant LG, Kuppen PJK, C RF, Ferrone S. Differential role of HLA-A and HLA-B, C expression levels as prognostic markers in colon and rectal cancer. J Immunother Cancer 2022, 10.
Menon AG, Morreau H, Tollenaar RA, Alphenaar E, Van Puijenbroek M, Putter H, Janssen-Van Rhijn CM, Van De Velde CJ, Fleuren GJ, Kuppen PJ. Down-regulation of HLA-A expression correlates with a better prognosis in Colorectal cancer patients. Lab Invest. 2002;82:1725–33.
Bijen CB, Bantema-Joppe EJ, de Jong RA, Leffers N, Mourits MJ, Eggink HF, van der Zee AG, Hollema H, de Bock GH, Nijman HW. The prognostic role of classical and nonclassical MHC class I expression in endometrial cancer. Int J Cancer. 2010;126:1417–27.
Leffers N, Gooden MJ, Mokhova AA, Kast WM, Boezen HM, Ten Hoor KA, Hollema H, Daemen T, van der Zee AG, Nijman HW. Down-regulation of proteasomal subunit MB1 is an Independent predictor of improved survival in Ovarian cancer. Gynecol Oncol. 2009;113:256–63.
van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, Andre P, et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer vaccines. Cell. 2018;175:1744.
Borst L, van der Burg SH, van Hall T. The NKG2A-HLA-E Axis as a Novel checkpoint in the Tumor Microenvironment. Clin Cancer Res. 2020;26:5549–56.
Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-Cell transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–62.
Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–7.
Giroux Leprieur E, Helias-Rodzewicz Z, Takam Kamga P, Costantini A, Julie C, Corjon A, Dumenil C, Dumoulin J, Giraud V, Labrune S et al. Sequential ctDNA whole-exome sequencing in advanced lung adenocarcinoma with initial durable Tumor response on immune checkpoint inhibitor and late progression. J Immunother Cancer 2020, 8.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 blockade in Melanoma. N Engl J Med. 2016;375:819–29.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Rodig SJ, Gusenleitner D, Jackson DG, G**i E, Giobbie-Hurder A, ** C, Chang H, Lovitch SB, Horak C, Weber JS et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic Melanoma. Sci Transl Med. 2018;10.
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. Impaired HLA Class I Antigen Processing and Presentation as a mechanism of Acquired Resistance to Immune checkpoint inhibitors in Lung Cancer. Cancer Discov. 2017;7:1420–35.
Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, **e L, Huang Q, Li F, Li CY. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693–8.
Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, et al. Suppression of type I IFN Signaling in Tumors mediates resistance to Anti-PD-1 treatment that can be overcome by Radiotherapy. Cancer Res. 2017;77:839–50.
Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in Breast cancer cells. PLoS ONE. 2012;7:e32542.
Srivastava RM, Trivedi S, Concha-Benavente F, Hyun-Bae J, Wang L, Seethala RR, Branstetter BFt, Ferrone S, Ferris RL. STAT1-Induced HLA Class I Upregulation enhances immunogenicity and clinical response to Anti-EGFR mAb Cetuximab Therapy in HNC patients. Cancer Immunol Res. 2015;3:936–45.
Liang YH, Chen KH, Tsai JH, Cheng YM, Lee CC, Kao CH, Chan KY, Chen YT, Hsu WL, Yeh KH. Proteasome inhibitors restore the STAT1 pathway and enhance the expression of MHC class I on human colon Cancer cells. J Biomed Sci. 2021;28:75.
Adachi Y, Kamiyama H, Ichikawa K, Fukushima S, Ozawa Y, Yamaguchi S, Goda S, Kimura T, Kodama K, Matsuki M, et al. Inhibition of FGFR reactivates IFNgamma Signaling in Tumor cells to enhance the combined antitumor activity of Lenvatinib with Anti-PD-1 antibodies. Cancer Res. 2022;82:292–306.
Watanabe S, Hayashi H, Haratani K, Shimizu S, Tanizaki J, Sakai K, Kawakami H, Yonesaka K, Tsurutani J, Togashi Y, et al. Mutational activation of the epidermal growth factor receptor down-regulates major histocompatibility complex class I expression via the extracellular signal-regulated kinase in non-small cell Lung cancer. Cancer Sci. 2019;110:52–60.
Kang SH, Keam B, Ahn YO, Park HR, Kim M, Kim TM, Kim DW, Heo DS. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology. 2019;8:e1515057.
Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116:180–3.
Kobayashi M, Fujiyama N, Tanegashima T, Narita S, Yamamoto Y, Fujimoto N, Ueda S, Takeuchi A, Numakura K, Habuchi T, et al. Effect of HLA genotype on intravesical recurrence after bacillus Calmette-Guerin therapy for non-muscle-invasive Bladder cancer. Cancer Immunol Immunother. 2022;71:727–36.
Rouanne M, Adam J, Radulescu C, Letourneur D, Bredel D, Mouraud S, Goubet AG, Leduc M, Chen N, Tan TZ et al. BCG therapy downregulates HLA-I on malignant cells to subvert antitumor immune responses in Bladder cancer. J Clin Invest. 2022;132.
Kiselyov A, Bunimovich-Mendrazitsky S, Startsev V. Treatment of non-muscle invasive Bladder cancer with Bacillus Calmette-Guerin (BCG): Biological markers and simulation studies. BBA Clin. 2015;4:27–34.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with Melanoma. Nature. 2017;547:217–21.
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–6.
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, **ong F, Guo C, Wu X, Li Y, Li X, et al. Neoantigen vaccine: an emerging Tumor immunotherapy. Mol Cancer. 2019;18:128.
Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA database. Nucleic Acids Res. 2020;48:D948–55.
Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–78.
Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–29.
Gigoux M, Holmstrom MO, Zappasodi R, Park JJ, Pourpe S, Bozkus CC, Mangarin LMB, Redmond D, Verma S, Schad S, et al. Calreticulin mutant myeloproliferative Neoplasms induce MHC-I skewing, which can be overcome by an optimized peptide cancer vaccine. Sci Transl Med. 2022;14:eaba4380.
Lhotakova K, Grzelak A, Polakova I, Vackova J, Smahel M. Establishment and characterization of a mouse Tumor cell line with irreversible downregulation of MHC class I molecules. Oncol Rep. 2019;42:2826–35.
Cao F, Yan M, Liu Y, Liu L, Ma G. Photothermally controlled MHC class I restricted CD8(+) T-Cell responses elicited by Hyaluronic Acid Decorated Gold nanoparticles as a vaccine for Cancer Immunotherapy. Adv Healthc Mater. 2018;7:e1701439.
Jung K, Son MJ, Lee SY, Kim JA, Ko DH, Yoo S, Kim CH, Kim YS. Antibody-mediated delivery of a viral MHC-I epitope into the cytosol of target Tumor cells repurposes virus-specific CD8(+) T cells for cancer immunotherapy. Mol Cancer. 2022;21:102.
Millar DG, Ramjiawan RR, Kawaguchi K, Gupta N, Chen J, Zhang S, Nojiri T, Ho WW, Aoki S, Jung K, et al. Antibody-mediated delivery of viral epitopes to tumors harnesses CMV-specific T cells for cancer therapy. Nat Biotechnol. 2020;38:420–5.
Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–9.
Zhu J, Ke Y, Liu Q, Yang J, Liu F, Xu R, Zhou H, Chen A, **ao J, Meng F, et al. Engineered Lactococcus lactis secreting Flt3L and OX40 ligand for in situ vaccination-based cancer immunotherapy. Nat Commun. 2022;13:7466.
Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A, et al. Personalized RNA neoantigen vaccines stimulate T cells in Pancreatic cancer. Nature. 2023;618:144–50.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66.
Marofi F, Achmad H, Bokov D, Abdelbasset WK, Alsadoon Z, Chupradit S, Suksatan W, Shariatzadeh S, Hasanpoor Z, Yazdanifar M, et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res Ther. 2022;13:140.
Sanchez-Paulete AR, Mateus-Tique J, Mollaoglu G, Nielsen SR, Marks A, Lakshmi A, Khan JA, Wilk CM, Pia L, Baccarini A, et al. Targeting macrophages with CAR T cells delays solid Tumor progression and enhances Antitumor Immunity. Cancer Immunol Res. 2022;10:1354–69.
Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-Based CAR T cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018;78:1031–43.
DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, Sadelain M. Low-dose Radiation Conditioning enables CAR T cells to mitigate Antigen Escape. Mol Ther. 2018;26:2542–52.
Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–44.
Zhang S, Kohli K, Black RG, Yao L, Spadinger SM, He Q, Pillarisetty VG, Cranmer LD, Van Tine BA, Yee C, et al. Systemic Interferon-gamma increases MHC Class I expression and T-cell infiltration in Cold tumors: results of a phase 0 clinical trial. Cancer Immunol Res. 2019;7:1237–43.
Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19:237–53.
Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. 2015;12:527–40.
Kamrava M, Bernstein MB, Camphausen K, Hodge JW. Combining radiation, immunotherapy, and antiangiogenesis agents in the management of cancer: the three musketeers or just another quixotic combination? Mol Biosyst. 2009;5:1262–70.
Liauw SL, Connell PP, Weichselbaum RR. New paradigms and future challenges in radiation oncology: an update of biological targets and technology. Sci Transl Med. 2013;5:173sr172.
Zebertavage LK, Alice A, Crittenden MR, Gough MJ. Transcriptional upregulation of NLRC5 by Radiation drives STING- and Interferon-Independent MHC-I expression on Cancer cells and T cell cytotoxicity. Sci Rep. 2020;10:7376.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71.
Zhou Y, Bastian IN, Long MD, Dow M, Li W, Liu T, Ngu RK, Antonucci L, Huang JY, Phung QT et al. Activation of NF-kappaB and p300/CBP potentiates cancer chemoimmunotherapy through induction of MHC-I antigen presentation. Proc Natl Acad Sci U S A. 2021;118.
Newcomb EW, Demaria S, Lukyanov Y, Shao Y, Schnee T, Kawashima N, Lan L, Dewyngaert JK, Zagzag D, McBride WH, Formenti SC. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin Cancer Res. 2006;12:4730–7.
de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20:5384–91.
Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy broadens the range of Tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol Immunother. 2012;61:2343–56.
Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–23.
He S, Yin T, Li D, Gao X, Wan Y, Ma X, Ye T, Guo F, Sun J, Lin Z, Wang Y. Enhanced interaction between natural killer cells and Lung cancer cells: involvement in gefitinib-mediated immunoregulation. J Transl Med. 2013;11:186.
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang SY, Hopson C, et al. The BRAF and MEK inhibitors Dabrafenib and Trametinib: effects on Immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639–51.
Khallouf H, Marten A, Serba S, Teichgraber V, Buchler MW, Jager D, Schmidt J. 5-Fluorouracil and interferon-alpha immunochemotherapy enhances immunogenicity of murine Pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J Immunother. 2012;35:245–53.
Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, Vimond N, Concha A, Garrido F, Isambert N, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013;73:3499–510.
Wang X, Waschke BC, Woolaver RA, Chen SMY, Chen Z, Wang JH. MHC class I-independent activation of virtual memory CD8 T cells induced by chemotherapeutic agent-treated cancer cells. Cell Mol Immunol. 2021;18:723–34.
Acknowledgements
The authors expressed their profound gratitude to every lab member. The schematic diagrams were created by Figdraw.
Funding
This work was supported by the National Natural Science Foundation of China (82172691 and 81772710) and Nan**g Science and Technology Development Key Project (YKK19011).
Author information
Authors and Affiliations
Contributions
Rong Yang and Hongqian Guo designed this study. **angyu Wu and Tianhang Li wrote this paper and drew the figures. Rui Jiang and **n Yang collected related references and made the table. Each author reviewed the final version of article before giving their approval for publication. **angyu Wu and Tianhang Li made equal contributions to the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, X., Li, T., Jiang, R. et al. Targeting MHC-I molecules for cancer: function, mechanism, and therapeutic prospects. Mol Cancer 22, 194 (2023). https://doi.org/10.1186/s12943-023-01899-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01899-4