
Abstract
Background
Currently, only limited knowledge is available regarding the phenotypic association between fibroblast growth factor receptor 3 (FGFR3) alterations and the tumor microenvironment (TME) in bladder cancer (BLCA).
Methods
A multi-omics analysis on 389 BLCA and 35 adjacent normal tissues from a cohort of OMPU-NCC Consortium Japan was retrospectively performed by integrating the whole-exome and RNA-sequence dataset and clinicopathological record. A median follow-up duration of all BLCA cohort was 31 months.
Results
FGFR3 alterations (aFGFR3), including recurrent mutations and fusions, accounted for 44% of non-muscle invasive bladder cancer (NMIBC) and 15% of muscle-invasive bladder cancer (MIBC). Within MIBC, the consensus subtypes LumP was significantly more prevalent in aFGFR3, whereas the Ba/Sq subtype exhibited similarity between intact FGFR3 (iFGFR3) and aFGFR3 cases. We revealed that basal markers were significantly increased in MIBC/aFGFR3 compared to MIBC/iFGFR3. Transcriptome analysis highlighted TIM3 as the most upregulated immune-related gene in iFGFR3, with differential immune cell compositions observed between iFGFR3 and aFGFR3. Using EcoTyper, TME heterogeneity was discerned even within aFGFR cases, suggesting potential variations in the response to checkpoint inhibitors (CPIs). Among 72 patients treated with CPIs, the objective response rate (ORR) was comparable between iFGFR3 and aFGFR3 (20% vs 31%; p = 0.467). Strikingly, a significantly higher ORR was noted in LumP/aFGFR3 compared to LumP/iFGFR3 (50% vs 5%; p = 0.022). This trend was validated using data from the IMvigor210 trial. Additionally, several immune-related genes, including IDO1, CCL24, IL1RL1, LGALS4, and NCAM (CD56) were upregulated in LumP/iFGFR3 compared to LumP/aFGFR3 cases.
Conclusions
Differential pathways influenced by aFGFR3 were observed between NMIBC and MIBC, highlighting the upregulation of both luminal and basal markers in MIBC/aFGFR3. Heterogeneous TME was identified within MIBC/aFGFR3, leading to differential outcomes for CPIs. Specifically, a favorable ORR in LumP/aFGFR3 and a poor ORR in LumP/iFGFR3 were observed. We propose TIM3 as a potential target for iFGFR3 (ORR: 20%) and several immune checkpoint genes, including IDO1 and CCL24, for LumP/iFGFR3 (ORR: 5%), indicating promising avenues for precision immunotherapy for BLCA.
Similar content being viewed by others


Introduction
BLCA poses a significant global health challenge, ranking as the fourth most common cancer and the eighth leading cause of cancer-related deaths in men [1]. The introduction of immune checkpoint inhibitors (CPIs) in BLCA treatment, sanctioned by the Food and Drug Administration (FDA) in 2017, marked a pivotal shift in therapeutic strategies [2]. However, the efficacy of CPIs remains limited, with the majority of patients showing minimal or no response. Patients with progressive disease (PD) at their best overall response accounted for 48.5% with no survival benefit compared to the second-line chemotherapy [3]. Therefore, understanding the molecular intricacies governing CPI response and exploring innovative methods to enhance CPI effectiveness are imperative.
Genetic alterations in fibroblast growth factor receptor 3 (FGFR3) are frequently identified in BLCA [4, 5]. Previous studies have linked FGFR3 alterations (aFGFR3) to luminal papillary tumors characterized by diminished T-cell infiltrations [6]. While initial assumptions suggested reduced sensitivity of aFGFR3 tumors to CPIs, recent research contradicts this notion, demonstrating comparable CPI effects between aFGFR3 and intact FGFR3 (iFGFR3) BLCA cases [7, 8]. Consequently, exploring the tumor microenvironment (TME) in the context of FGFR3 status emerges as a promising avenue for novel therapeutic interventions. To address this, we conducted a comprehensive multi-omics analysis encompassing 389 BLCA cases and 35 adjacent normal tissues, aiming to unravel the intricate relationship between aFGFR3 and the TME.
Results
FGFR Alterations in NMIBC and MIBC
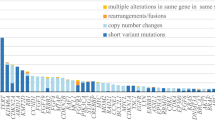
The cohort in the present study included 124 non-muscle invasive bladder cancer (NMIBC: 5-year overall survival (OS) rate: 83%) and 265 muscle-invasive bladder cancer (MIBC: 5-year OS rate: 35%) with a median follow-up of 36 and 22 months, respectively (Supplementary Fig. 1A). We first assessed mRNA expression levels of FGFR family genes in the cohort. FGFR3 is actively transcribed among the FGFR families in the bladder, especially in BLCA (Supplementary Fig. 1B). FGFR3 was the most frequently mutated gene (81/389; 21%) among the FGFR families including FGFR1 (19/389; 5%), FGFR2 (8/389; 2%), and FGFR4 (12/389; 3%) (Supplementary Fig. 1C). A previous TCGA publication defined 58 significantly mutated genes (SMGs) in BLCA [9], which was largely recapitulated in our cohort including the clinicopathological characteristics (Fig. 1A, B).

FGFR Alterations in NMIBC and MIBC. A Mutation landscape of 58 significantly mutated genes defined by the TCGA publication [9] in 389 bladder cancer (BLCA) samples from the OMPU-NCC cohort. The patients were classified into pTa (n = 59), pT1 (n = 65), and ≥ pT2 (n = 265, MIBC: muscle-invasive bladder cancer). B Recurrent mutation rate of 58 significantly mutated genes according to pathological T stages. C Schematic of the FGFR3 fusions identified in our cohort. FGFR3-TACC3 fusions were found in 14 of 289 patients, and the most frequent pattern (7 of 11) is shown. NSD2 and SPON2 are newly identified fusion partners. D FGFR3 mRNA expression levels according to the FGFR3 alterations. The difference was assessed by the Mann–Whitney U test; p < 0.05*, p < 0.001**, p < 0.0001***. E, F Kaplan–Meier curves demonstrating progression-free survival (PFS) in non-muscle-invasive bladder cancer (NMIBC) (E) and overall survival (OS) in MIBC (F). A log-rank test was used to assess the survival difference between the two groups; p < 0.05*
To delineate the allelic difference in FGFR3 among ethnicity, we sought to assess whether there is a specific allelic variant in the germline for the Asian population. GnomADv3.0, an integrative germline dataset of 71,702 individuals (mostly Western population), was utilized for the control [10]. We referred the Asian germline dataset (jMorp-14KJPN) [11] and identified five significantly enriched non-synonymous single nucleotide polymorphisms (SNPs) on the FGFR3 gene locus that are specific to the Asian population (Q29H, G65R, L164V, T450M, and A720S) (Supplementary Fig. 1D). However, these SNPs were not enriched in BLCA samples (Supplementary Table 1). Compared to BLCA with iFGFR3, FGFR3 mRNA expression level was significantly upregulated in patients with recurrent FGFR3 mutations but not in patients with the SNPs (Supplementary Fig. 1E). There seemed to be no survival difference based on the FGFR3 status (Supplementary Fig. 1F), indicating no clinical implication of these Asian-specific SNPs in FGFR3.
FGFR3 mutations was predominantly observed in cases with lower malignant properties such as NMIBC (pTa: 51%, pT1: 29%, more than pT2: 12%), low grade and negative lymph vascular invasions (Table 1). Regarding the mutational alleles, the TCGA publication, which only consists of MIBC samples, reports S249C and Y373C as the top two frequent FGFR3 mutations in BLCA (Supplementary Fig. 2A). We noted that recurrent K650E and T757P nonsynonymous mutations at the kinase domain (KD) were frequently observed in MIBC in our cohort compared to the TCGA cohort (Supplementary Fig. 2B). Although we examined the prognosis of five MIBC cases with mutation at KD (three in K650E and two in T757P), there was no difference in OS compared to that in cases with other FGFR3 alterations (Supplementary Fig. 2C). Interestingly, we found that FGFR3 mutations at KD were more prevalent in MIBC than in NMIBC cases (p = 0.021) (Supplementary Fig. 2D).
FGFR3 mRNA expression levels were consistently upregulated in aFGFR3 compared to iFGFR3, regardless of the mutation sites (Supplementary Fig. 2E). The present study exhibited a frequency of 4% (17/389) for FGFR3 fusions (Table 1), including novel fusion partners (NSD2 and SPON2) (Fig. 1C). No histological variant was observed in cases with FGFR3 fusions (Table 1). The KD located at the C-terminus of FGFR3 has been retained in 13 of 17 (77%) fusions, and the mRNA expression level was significantly upregulated in cases with FGFR3 fusions compared to cases with iFGFR3 (Supplementary Fig. 2F). Upon stratifying our cohort into NMIBC and MIBC categories, FGFR3 mRNA expression levels were significantly higher in aFGFR3 cases than in iFGFR3 cases in both NMIBC and MIBC (Fig. 1D), with the highest median mRNA expression levels observed in MIBC patients with FGFR3 fusions. This finding underscores the clinical importance of detecting FGFR3 fusions, alongside mutations, in advanced MIBC patients and accentuates the importance of considering recently approved FGFR3 inhibitors [12]. The FGFR3 protein expression levels were increased in aFGFR3 compared to iFGFR3 cases (Supplementary Fig. 2G, H). We investigated the progression free survival (PFS) of 124 NMIBC patients (Fig. 1E). Patients with recurrent FGFR3 mutations showed a significantly better PFS compared to those with iFGFR3 (p = 0.037). However, this distinction was not evident in patients with FGFR3 fusions (p = 0.806). In the context of OS among 265 MIBC patients, no significant differences were observed based on FGFR3 status (Fig. 1F).
The Association between FGFR3 alteration and molecular Subtypes
We have adopted the established consensus MIBC subtype [6], the UROMOL subtype for NMIBC [13], and Baylor college subtype [3B-3D). In the overall cohort (n = 389), FGFR3 alterations were enriched in class_1 (54%) and class_3 (94%) for the UROMOL subtype and LumP (42%) for consensus MIBC subtype (Fig. 2B and Supplementary Fig. 3E). An elevated FGFR3 mRNA expression level was confirmed within these molecular subtypes (Fig. 2C, D and Supplementary Fig. 3F).

Association between FGFR3 Alteration and Molecular Subtypes. A Summary of FGFR3 alterations, FGFR3 mRNA expression, histological variant, consensus MIBC subtypes [6], UROMOL NMIBC subtypes [13], and Baylor college [https://github.com/Genomon-Project/GenomonFisher). The software included in GenomonPipeline was used not only to analyze cancer samples, but also to compare tumor samples, for which paired normal sample were available, in pairs with normal samples to detect somatic mutations more accurately. In addition, false-positive somatic mutations from cancer genome sequencing data were filtered by Genomon Mutation Filter (https://github.com/Genomon-Project/GenomonMutationFilter). Finally, annotating process for the filtered mutations list was organized by Genomon Mutation Annotator (https://github.com/Genomon-Project/GenomonMutationAnnotator) and ANNOVAR:20,210,202 [26]. These annotations included information on amino acid changes and allele frequencies in several public databases. For publicly available dataset of germline whole-genome sequencing, gnomADv3.0 (https://gnomad.broadinstitute.org/news/2019-10-gnomad-v3-0/) [10] and jMorp-14KJPN (https://jmorp.megabank.tohoku.ac.jp/202112/) [11] were utilized to filter out false positive somatic mutations in the analysis of single nucleotide polymorphisms (SNPs). The mutation landscape was visualized using a script based on CoMut library(https://github.com/vanallenlab/comut). Lolipop mutation plots were generated by using Matplotlib. The script also acquired domain and motif features from UniProt database via Proteins API. < < # The proteins API: accessing key integrated protein and genome information (https://doi.org/10.1093/nar/gkx237)> >.
For the RNA-seq analysis, STAR:2.5.2a (https://github.com/alexdobin/STAR) was used for the map** of FASTQ on GRCh38. Then, featureCounts (SUBREAD): 2.0.1 (http://subread.sourceforge.net/) was adopted to count the number of reads mapped on exon regions by gene symbol. Raw read counts were used after normalization to TPM (transcripts per million). Regarding the molecular subtypes, we utilized the consensus MIBC subtype (https://github.com/cit-bioinfo/consensusMIBC) [6], the UROMOL subtype (https://github.com/sialindskrog/classifyNMIBC) [13], and the Baylor college subtype [14]. The detection and visualization process of FGFR3-fusion transcript was performed using Arriba platform (https://github.com/suhrig/arriba). Estimated immune-related cell composition was calculated by CIBERSORT [27]. For the deconvolution of tumor microenvironment from the bulk RNA-seq data, EcoTyper (https://github.com/digitalcytometry/ecotyper) was adopted [16]. For differentially expressed gene (DEG) analysis, the DEseq2 platform (https://lashlock.github.io/compbio/R_presentation.html) was conducted using a raw read count matrix from the present cohort.
For publicly available datasets, TCGA data set was analyzed using the cBio Cancer Genomics Portal (cBioPortal; www.cbioportal.org). The raw data from the IMvigor210 trial [17] was downloaded from (http://research-pub.gene.com/IMvigor210CoreBiologies/). Heatmaps were created using Morpheus (https://software.broadinstitute.org/morpheus/).
Immunohistochemistry
PD-L1 protein expression in immunohistochemistry (IHC) was evaluated in tumor samples obtained from patients using the PD-L1 IHC 22C3 pharmDx assay (Agilent Technologies, Santa Clara, CA) and the 22C3 anti–PD-L1 antibody (Merck & Co., Kenilworth, NJ) [28]. The Combined Positive Score (CPS) method was employed to determine PD-L1 protein expression. This approach quantifies the number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) and divides it by the total number of viable tumor cells, then multiplies the result by 100. Immunohistochemical staining was conducted using the Discovery ULTRA System (Roche Diagnostics, Basel, Switzerland) as per the manufacturer's guidelines. A panel of antibodies was employed to evaluate the immune profile of the tumor samples, including TIM-3 (rabbit monoclonal antibody, D5D5R, Cell Signaling Technology, Danvers, MA, USA; diluted 1:200), CD8 (monoclonal mouse clone, C8/144B, DAKO; diluted 1:200), and FOXP3 (mouse monoclonal clone, 236A/E7, Abcam; diluted 1:100). At least two researchers independently assessed the immunohistochemistry results to ensure accuracy and reproducibility. The criteria for determining positive cell count were as follows: membrane staining of any intensity for TIM-3 and CD8, or nuclear staining for FOXP3 on ≥ 1% of cells at a high-power field. In the clinical samples, FGFR3 protein expression was evaluated using an FGFR3 rabbit monoclonal antibody (MA5-32,620, ThermoFisher Scientific). The H-score, ranging from 0 to 300, was calculated as (3 × percentage of strongly staining nuclei + 2 × percentage of moderately staining nuclei + percentage of weakly staining nuclei), allowing for a semi-quantitative assessment of protein expression levels. The CPS of PD-L1 and H-score of FGFR3 were evaluated by two board-certified pathologists to provide a robust and reliable foundation for further data analysis and interpretation in the context of molecular pathology.
Conclusions
We comprehensively investigated the biological implication of aFGFR3 in BLCA. Differential pathways were affected by aFGFR3 between NMIBC and MIBC, particularly emphasizing the significant upregulation of both luminal and basal markers in MIBC/aFGFR3 cases. Crucially, our study underscores the heterogeneous nature of the TME within MIBC/aFGFR3, leading to differential treatment outcomes for CPIs. In particular, favorable ORR in LumP/aFGFR3 and poor ORR in LumP/iFGFR3 were noted. We propose TIM3 for iFGFR3 (ORR: 20% in our cohort) and several immune checkpoint genes for LumP/iFGFR3 (ORR: 5% in our cohort), including IDO1, CCL24, IL1RL1, LGALS4, and NCAM (CD56) as potential druggable targets. These findings offer promising avenues for future precision immunotherapy, indicating a plausible direction for enhancing treatment outcomes in BLCA patients.
Availability of data and materials
The raw sequencing data and clinical data can be obtained from the Osaka Medical and Pharmaceutical University Translational Research Program Biobank (OMPU-TR Biobank) (https://www.ompu.ac.jp/department/rdcenter/transregular/); however, restrictions apply to the availability of these data, and they are not publicly accessible. Data are available to National Cancer Center Research Institute and OMPU investigators and their external affiliates, including academic and commercial partners, provided that they have approval from the Institutional Review Board (IRB) and a data use agreement. Samples and data shared with external entities must be de-identified. Any additional information required to reanalyze the data reported in this work is available from the Lead Contact (KK; kazumasa.komura@ompu.ac.jp, AY; ayoshimi@ncc.go.jp) upon request.
Abbreviations
- BLCA:
-
Bladder cancer
- CE:
-
Cellular ecotypes
- CI:
-
Confidence interval
- CPIs:
-
Checkpoint inhibitors
- CPS:
-
Combined positive score
- CR:
-
Complete response
- CSS:
-
Cancer-specific survival
- EMT:
-
Epithelial-mesenchymal transition
- FGFR3:
-
Fibroblast growth factor receptor 3
- GSEA:
-
Gene set enrichment analysis
- KD:
-
Kinase domain
- MIBC:
-
Muscle invasive bladder cancer
- NMIBC:
-
Non-muscle invasive bladder cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PD:
-
Progressive disease
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- SD:
-
Stable disease
- SNPs:
-
Single nucleotide polymorphisms
- TMB:
-
Tumor mutation burden
- TME:
-
Tumor microenvironment
- UC:
-
Urothelial carcinoma
- WES:
-
Whole exome sequencing
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. 2017;376:1015–26.
Fradet Y, Bellmunt J, Vaughn DJ, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, Necchi A, et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: results of >2 years of follow-up. Ann Oncol. 2019;30:970–6.
Thomas J, Sonpavde G. Molecularly Targeted Therapy towards Genetic Alterations in Advanced Bladder Cancer. Cancers (Basel). 2022;14:1795.
Knowles MA. FGFR3 – a Central Player in Bladder Cancer Pathogenesis? Bladder Cancer. 2020;6:403–23.
Kamoun A, de Reynies A, Allory Y, Sjodahl G, Robertson AG, Seiler R, Hoadley KA, Groeneveld CS, Al-Ahmadie H, Choi W, et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur Urol. 2020;77:420–33.
Rose TL, Weir WH, Mayhew GM, Shibata Y, Eulitt P, Uronis JM, Zhou M, Nielsen M, Smith AB, Woods M, et al. Fibroblast growth factor receptor 3 alterations and response to immune checkpoint inhibition in metastatic urothelial cancer: a real world experience. Br J Cancer. 2021;125:1251–60.
Wang L, Gong Y, Saci A, Szabo PM, Martini A, Necchi A, Siefker-Radtke A, Pal S, Plimack ER, Sfakianos JP, et al. Fibroblast Growth Factor Receptor 3 Alterations and Response to PD-1/PD-L1 Blockade in Patients with Metastatic Urothelial Cancer. Eur Urol. 2019;76:599–603.
Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171(540–556): e525.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Takayama J, Tadaka S, Yano K, Katsuoka F, Gocho C, Funayama T, Makino S, Okamura Y, Kikuchi A, Sugimoto S, et al. Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. Nat Commun. 2021;12:226.
Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2019;381:338–48.
Lindskrog SV, Prip F, Lamy P, Taber A, Groeneveld CS, Birkenkamp-Demtroder K, Jensen JB, Strandgaard T, Nordentoft I, Christensen E, et al. An integrated multi-omics analysis identifies prognostic molecular subtypes of non-muscle-invasive bladder cancer. Nat Commun. 2021;12:2301.
Mo Q, Nikolos F, Chen F, Tramel Z, Lee YC, Hayashi K, **ao J, Shen J, Chan KS. Prognostic Power of a Tumor Differentiation Gene Signature for Bladder Urothelial Carcinomas. J Natl Cancer Inst. 2018;110:448–59.
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37:773–82.
Luca BA, Steen CB, Matusiak M, Azizi A, Varma S, Zhu C, Przybyl J, Espin-Perez A, Diehn M, Alizadeh AA, et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell. 2021;184:5482-5496 e5428.
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20.
Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res. 2016;4:563–8.
Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, Plimack ER, Vaena D, Grimm MO, Bracarda S, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–22.
Wang L, Saci A, Szabo PM, Chasalow SD, Castillo-Martin M, Domingo-Domenech J, Siefker-Radtke A, Sharma P, Sfakianos JP, Gong Y, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun. 2018;9:3503.
Falchook GS, Ribas A, Davar D, Eroglu Z, Wang JS, Luke JJ, Hamilton EP, Di Pace B, Wang T, Ghosh S, et al. Phase 1 trial of TIM-3 inhibitor cobolimab monotherapy and in combination with PD-1 inhibitors nivolumab or dostarlimab (AMBER). J Clin Oncol. 2022;40:2504–2504.
Tang K, Wu YH, Song Y, Yu B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol. 2021;14:68.
Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2020;9:1777625.
Brian SW, Aurélien de R, Aaron MN, Joshua JW, Andrew L, Florent P, Alberto V, Yating L, Haojun L, Xu X, et al. Community assessment of methods to deconvolve cellular composition from bulk gene expression. bioRxiv. 2022.06.03.494221. https://doi.org/10.1101/2022.06.03.494221.
Yachie N, Natsume T. Robotic crowd biology with Maholo LabDroids. Nat Biotechnol. 2017;35:310–2.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38: e164.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Balar AV, Castellano D, O’Donnell PH, Grivas P, Vuky J, Powles T, Plimack ER, Hahn NM, de Wit R, Pang L, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:1483–92.
Acknowledgements
The authors wish to acknowledge Yuriko Kawakami, Akiko Kagotani, Sayaka Sasada, and Rintaro Oide in the Translational Research Program of Osaka Medical and Pharmaceutical University for their help in processing the clinical samples and develo** the experimental models in the present study.
Funding
K.K. was partially supported by Grant-in-Aid No. 21H03070 (Japan Society for the Promotion of Science: JSPS), the Kenzo Suzuki Memorial Foundation, the SGH Foundation, the Naito Memorial Foundation, and the Cancer Translational Research Foundation of Japanese Urological Association (JUA). A.Y. was partly supported by funding from JSPS KAKENHI (Grant-in-Aid No. 21H04828), the Japan Agency for Medical Research and Development (AMED; grant number JP22jm0210085, JP22am0401007, and JP22ym0126804 (21-A-22)), National Cancer Center Research and Development Funds (grant number 2020-A-2 and 2023-S-6), and Takeda Science Foundation.
Author information
Authors and Affiliations
Contributions
KK: conceptualization, data curation, analysis, investigation, methodology, software, visualization, writing original draft, supervision. KH: data curation, analysis, investigation, methodology, software, visualization. ST: data curation, investigation, investigation, writing the original draft. TTu**o: analysis, investigation. KNishimura: investigation, validation. MI: validation, methodology. TH: methodology, validation. AU: methodology, validation. TO: data curation. SY: data curation, analysis. KNakamori: investigation. SK: data curation. RM: investigation. MA: investigation, validation. YY: data curation, analysis. TTakai: validation. TTsutsumi: validation, methodology. KTaniguchi: investigation. TTanaka: investigation, validation. KTakahara: conceptualization, investigation. TK: validation, methodology. TI: validation. YH: review and editing, supervision. FO: review and editing, supervision. YS: data analysis. AY: investigation, writing original draft, review and editing, supervision. HA: review and editing, supervision.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study has been performed based on the principles of the World Medical Association Declaration of Helsinki. The study designs were approved by the institutional review board at Osaka Medical and Pharmaceutical University [OMPU-IRB approval number: No. 2305-11 (approval date: Nov 24th, 2017), No. 2344-10 (approval date: Nov 24th, 2017), No. 2523-3 (approval date: Jul 27th, 2018), No. 2808-2 (approval date: Jan 10th, 2020), No. 2571-6 (approval date: Nov 7th, 2018)]) and National Cancer Center Japan (NCC-IRB approval number: 2020–486, Date of approval: February 9th, 2021). All patients provided written informed consent prior to the analysis.
Consent for publication
All the authors, by giving their consent to participate, have given their consent for publication.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 1.
(A) Kaplan–Meier curves for overall survival (OS) in non-muscle invasive bladder cancer (NMIBC) (upper panel: 124 patients) and muscle-invasive bladder cancer (MIBC) (lower panel: 265 patients). (B) Violin plots for mRNA expression levels (TPM: transcripts per million) of FGFR families (FGFR1-4) in normal (n=35) and tumor (n=389) tissues. (C) Violin plots for mRNA expression levels (TPM) in each FGFR family in 389 tumor samples according to the presence or absence of mutation. The difference was assessed by the Mann–Whitney U test. (D) Comparison of single nucleotide polymorphisms (SNPs) between GnomADv3.0, an integrative germline dataset of 71,702 individuals (mostly Western population) [10] and Japanese germline dataset (jMorp-14KJPN) of 28,258 allele number [11] within the FGFR3 gene locus (NM_000142). (E) FGFR3 mRNA expression levels in FGFR3 intact, FGFR3 SNPs, FGFR3 mutants in 389 bladder cancer (BLCA) patients (The difference in the expression level was assessed by the Mann–Whitney U test; *p<0.05, n.s: non-significant). (F) Kaplan–Meier curves for cancer-specific survival (CSS), OS and progression-free survival (PFS) in NMIBC (left panels: 124 patients) and MIBC (right panels: 265 patients) according to the FGFR3 genetic alternations. Log-rank test was utilized to examine the difference in survival.
Additional file 2: Supplementary Figure 2.
(A) Mutation plot of FGFR3 (NM_000142) in 408 TCGA bladder cancer cohort [9]. (B) Mutation plots of FGFR3 (NM_000142) for cases with NMIBC (n=124) and MIBC (n=265). (C) Kaplan-Meier curves for OS in MIBC (n=265) according to mutations at the kinase domain (KD). (D) Estimated proportion of mutations at KD in NMIBC and MIBC cases. (E) FGFR3 mRNA expression level among the nonsynonymous mutations and SNPs in 389 BLCA patients. (F) FGFR3 mRNA expression levels according to FGFR3 alterations in 389 BLCA samples. Four samples harboring both mutation and fusion were assigned to the fusion group. The difference in the FGFR3 mRNA was assessed by the Mann–Whitney U test (*p<0.05). (G) Pie charts of the H-score for FGFR3 according to FGFR3 status. Chi-square test was utilized to assess the difference. (H) Representative images of Immunohistochemistry for FGFR3. H-score was evaluated by (3 x percentage of strongly staining nuclei + 2 x percentage of moderately staining nuclei + percentage of weakly staining nuclei, giving a range of 0 to 300).
Additional file 3: Supplementary Figure 3.
(A) Hierarchical clustering for the 18 tumor differentiation classifier genes from Baylor College which define the two subgroups with distinct expression patterns [14]. (B) Estimated proportion of consensus MIBC subtypes [6] in NMIBC and MIBC cases. (C) Estimated proportion of UROMOL NMIBC subtypes [13] in NMIBC and MIBC cases. (D) Estimated proportion of Baylor college subtypes in NMIBC and MIBC cases. (E) Pie charts of FGFR3 alterations in each molecular subtype. Fisher’s exact test was utilized to assess the difference. (F) FGFR3 mRNA expression levels (transcripts per million: TPM) according to the Baylor college subtypes. The difference was assessed by the Mann–Whitney U test. (G) Gene set enrichment analysis in 389 BLCA (iFGFR3: 295 cases vs aFGFR3: 94 cases) plotting all human MSigDB collections (Hallmark, C1-8: 23734 gene sets) by false discovery rate q-value (FDR-q) and normalized enrichment score (NES). (H) Gene set enrichment analysis (GSEA) of “HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION”, and“HALLMARK_ALLOGRAFT_REJECTION” that were top 2 downregulated pathways in aFGFR3.
Additional file 4: Supplementary Figure 4.
(A) Representative images of immunohistochemistry for PD-L1 using the PD-L1 IHC 22C3 pharmDx assay (Agilent Technologies, Santa Clara, CA) and the 22C3 anti–PD-L1 antibody (Merck & Co., Kenilworth, NJ) [28]. The PD-L1 protein expression is determined by the Combined Positive Score (CPS), the number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumor cells multiplied by 100. Corresponding hematoxylin-eosin stain (HE stain) is shown in the upper series. (B) CD274 mRNA expression level according to the CPS. (C) Pie chart of the PD-L1 CPS score (364 of 389 tumors were evaluable). (D) Pie chart of the PD-L1 CPS score in aFGFR3 (n=84) and iFGFR3 (n=280). (E) Representative images of immunohistochemistry for CD8. The cell count was evaluated at 400x magnification. (F) Cell count of CD8+ cells for the stromal, intratumor, and total region with high power field (x400). Mann-Whitney U test was used to examine the difference. (G) Heatmap of putative immune checkpoint genes according to FGFR3 status. (H) mRNA expression (transcripts per million: TPM) of immune checkpoint genes between iFGFR3 (n=295) and aFGFR3 (n=94) (Mann-Whitney U test was used to examine the difference. * p<0.05, **p<0.01, *** p<0.001, **** p<0.0001).
Additional file 5: Supplementary Figure 5.
(A) Correlation between “estimated proportion of T-cell CD8 from CIBERSORTx” and “cell count of CD8+ positive cells in HPF” (left panel), and “T-cells regulatory” and cell count of FOXP3 positive cells in HPF” (right panel), respectively. (B,C) The estimated proportion of each immune cell type from CIBERSORTx comparing iFGFR3 and aFGFR3 in (B) NMIBC and (C) MIBC. Mann-Whitney U test was used to examine the difference. * p<0.05,** p<0.001, *** p<0.0001, n.s: non-significant. (D) Pearson correlation coefficient among the estimated proportion of immune-related cells in NMIBC/iFGFR3 (n=69) and NMIBC/aFGFR3 (n=55). (E) Pearson correlation coefficient among the estimated proportion of immune-related cells in MIBC/iFGFR3 (n=226) and MIBC/aFGFR3 (n=39). (F) Individual cell states in each of ten cell types from the EcoTyper analysis. Representative genes defining cell states are shown in each cell type.
Additional file 6: Supplementary Figure 6.
(A) Estimated proportion of treatment response to CPIs including pembrolizumab (n=60) and avelumab (n=12). (B) Estimated proportion of treatment response to CPIs among cellular ecotypes (CEs) defined by EcoTyper. (C) Correlation of tumor mutation burden (TMB) with PD-L1 combined positive score (CPS), TIM3 positive cell count in high power field, and FGFR3 mRNA expression level. (D) Estimated proportion of consensus MIBC subtypes among in IMvigor210 trial [17] (n=274). (E) The ORR in the IMvigor210 trial (n=274) and the present cohort (OMPU: n=72) treated with CPIs in LumU subtype (left panel) and other subtypes including LumNS, NE-like, and stromal-rich (right panel). Fisher’s exact test was performed to assess the difference of the ORR according to FGFR3 status. Note that the data from IMvigor210 does not include the information on FGFR3 fusions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Komura, K., Hirosuna, K., Tokushige, S. et al. The Impact of FGFR3 Alterations on the Tumor Microenvironment and the Efficacy of Immune Checkpoint Inhibitors in Bladder Cancer. Mol Cancer 22, 185 (2023). https://doi.org/10.1186/s12943-023-01897-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01897-6