
Abstract
As a nontraditional T-cell subgroup, γδT cells have gained popularity in the field of immunotherapy in recent years. They have extraordinary antitumor potential and prospects for clinical application. Immune checkpoint inhibitors (ICIs), which are efficacious in tumor patients, have become pioneer drugs in the field of tumor immunotherapy since they were incorporated into clinical practice. In addition, γδT cells that have infiltrated into tumor tissues are found to be in a state of exhaustion or anergy, and there is upregulation of many immune checkpoints (ICs) on their surface, suggesting that γδT cells have a similar ability to respond to ICIs as traditional effector T cells. Studies have shown that targeting ICs can reverse the dysfunctional state of γδT cells in the tumor microenvironment (TME) and exert antitumor effects by improving γδT-cell proliferation and activation and enhancing cytotoxicity. Clarification of the functional state of γδT cells in the TME and the mechanisms underlying their interaction with ICs will solidify ICIs combined with γδT cells as a good treatment option.
Similar content being viewed by others
Introduction
In recent years, many studies have shown that cytotoxic γδT cells have a strong killing ability toward autologous, allogeneic or xenogeneic tumor cells, and their concentration is often closely related to the clinical prognosis of patients [1,2,3,4,5]. In contrast to αβT cells, γδT cells can recognize antigens in a way that is not restricted by major histocompatibility complex (MHC) molecules [6], and they mediate the killing of target cells in various ways, participate in immunomodulation, and have a broader tumor cell killing spectrum [7, 8]. As the “bridge” between innate immunity and adaptive immunity as well as the first line of defense against tumors, γδT cells play an important role in the occurrence and development of tumors and can be used to evaluate the prognosis of patients [8,9,10]. Therefore, immunotherapy based on γδT cells has been studied in many kinds of tumors [153]. Therefore, it is very important to further characterize the interaction between γδT cells and BTNs.
BTN3A/CD277 is an indispensable molecule in γδT-cell activation. Administration of a mAbs that activated BTN3A1 can enhance the activation of γδT cells and their cytotoxic effects [63]. By adding anti-CD277 mAbs and knocking down CD277, Harly et al. showed that CD277 plays a unique role in the response of Vγ9Vδ2T cells induced by PAgs, and the mechanism of γδT-cell activation depended on CD277 and was based on TCR signal transduction [154]. Without any other stimulation (such as anti-CD3 mAbs), administration of the soluble mAb 20.1 targeting BTN3A/CD277 indirectly increased the antigen recognition of Vγ9Vδ2T cells and mimicked PAgs to induce complete activation of Vγ9Vδ2T cells, accompanied by activation of Ca2+ signal transduction, upregulation of CD69 expression and increased IFN-γ secretion [154, 155]. The anti-BTN3A mAb 20.1 combined with Vγ9Vδ2T-cell immunotherapy restored the proliferation and cytotoxicity of Vγ9Vδ2T cells in tumors, prevented the exhaustion of adoptively transferred Vγ9Vδ2T cells, improved the survival rate of animals and reduced the tumor burden in blood and bone marrow [152]. Aude De Gassart and coworkers [156] also developed a new humanized anti-BTN3A mAb, ICT01, that can specifically activate Vγ9Vδ2T cells and upregulate the proportion of CD69+ Vγ9Vδ2T cells in a concentration-dependent manner. ICT01 can induce the degranulation of Vγ9Vδ2T cells and promote the production of IFN-γ, TNF-α, IL-8, IL-1β, monocyte chemoattractant protein 1 (MCP-1) and other proinflammatory cytokines. In addition, after the application of ICT01, the tumor growth rate of mice was obviously slowed. Importantly, the mAbs had no obvious effect on normal cells, highlighting the potential clinical value of the anti-BTN3A mAb ICT01.
After blocking BTN proteins, the response of γδT cells to PAgs was blocked, and there was no longer a tumor cell killing effect. Yamashiro et al. found that the expression level of BTN3 was inversely correlated to the activity of lymphocytes, and administration of the mAb 232-5 lead to phosphorylation of the BTN3A3 molecule and transduction of negative signals; these effects caused CD4+ and CD8+ T cells to act like CD4 + CD25+ Tregs, accompanied by a decrease in cell proliferation and cytokine secretion [157]. However, the mAb 20.1 has no significant stimulatory or costimulatory effect on αβT cells [154], suggesting that BTN proteins may jointly inhibit effector T cells through negative signal transmission and that BTN3A3 may be a novel target [158]. In addition, although the mAb 103.2 does not affect the proliferation and activation of CD8+ T cells, it can inhibit the response of γδT cells induced by PAgs and even inhibit the degranulation and cytokine secretion of Vγ9Vδ2T cells, decreasing their antitumor effects. Audrey Benyamine and coworkers also demonstrated that the mAbs 103.2 and 108.5 targeting the BTN3A molecule can completely inhibit the tumor cell lysis mediated by Vγ9Vδ2T cells [65, 152]. In addition to BTN3A, the B30.2 domain of BTN2A is required for the response of γδT cells to PAgs. The expression of the BTN2A1/BTN3A1 complex can trigger the activation of Vγ9Vδ2TCR, and the expression of BTN2A1 in cancer cells is related to the cytotoxicity of Vγ9Vδ2T cells. Anti-BTN2A1 mAbs can significantly inhibit the degranulation of Vγ9Vδ2T cells, and the inhibition of Vγ9Vδ2T-cell cytotoxicity induced by the 7.48 mAb generates an effect similar to the real TME [159].
Targeting BTN family proteins is beneficial for restoring the antitumor function of γδT cells and promoting their interaction with other immune cells, and they exert their antitumor effects synergistically. It has been reported that melanoma cells can hijack the interaction between pDCs and γδT cells to escape immune control, which is manifested as dysfunction of BTN3A and impaired ability of γδT cells to regulate ICs [160]. Conformational change of BTN3A1 is the key event of PAg perception [161], and BTN2A1 is the key ligand that binds to the Vγ9+ TCR γ chain; it can directly bind to the germline coding region of the Vγ9 chain in Vγ9Vδ2TCR and simultaneously bind to BTN3A1 on the cell surface. The synergistic effect of BTN3A1 and BTN2A1 enhances the recognition of target cells by Vγ9Vδ2T cells and plays an important role in PAg perception [162,163,164,165,166]. BTN3A1 can also recognize the N-mannosylated oligosaccharide in the near-membrane domain of CD45 and anchor the CD45 dimer near TCR, which may physically block the interaction of TCR-peptide-MHC-I complexes (pMHC), thus effectively inhibiting the separation of CD45 molecules from immune synapses and ultimately inhibiting the functions of TCRs and αβT cells [167]. Therefore, CD277-specific antibodies can restore the effector activity of αβT cells and induce BTN3A activity to mediate the synergistic killing of BTN3A1+ tumor cells by αβT cells and γδT cells in a BTN2A1-dependent manner. In summary, BTN family proteins are potential targets for fully exploiting the potential of γδT cells in IC therapy (Fig. 4).

The activation of γδT cells can be modulated by anti-BTN3A antibodies. BTN2A can bind to the γ chain of γδTCR and plays an important role with BTN3A in the activation of γδT cells by phosphoantigens. After the binding of an antagonistic monoclonal antibody to BTN3A, the activation of Vγ9Vδ2T cells will be blocked, and the cytotoxicity of Vγ9Vδ2T cells will decrease or even disappear. In contrast, after the use of an agonist targeting BTN3A, Vγ9Vδ2T cells have increased antigen sensitivity and enhanced ability to kill tumor cells, suggesting that targeting BTN family proteins can significantly regulate the immune response of γδT cells against tumor cells
Blocking inhibitory IC signaling can reverse the immunosuppressive state of γδT cells
ICIs can restore the proliferation and activation of γδT cells
The proliferation and activation of γδT cells were improved after administration of mAbs against ICs (Fig. 5). ICIs targeting B7-H3 and TIM-3 can improve the proliferation and/or activation of dysfunctional γδT cells. In colorectal cancer, knocking down or blocking B7-H3 can inhibit the apoptosis of Vδ2T cells, promote the proliferation of Vδ2T cells and induce the expression of the activation markers CD25 and CD69 in Vδ2T cells [168]. Caspase-3 participates in the activation of the TIM-3 signaling pathway, so γδT cells with upregulated TIM-3 are more prone to experience early apoptosis. After blocking TIM-3, the proliferation of Vγ9Vδ2T cells, the STAT phosphorylation level and the induction of IL-21 increased significantly [97, 169]. However, the expression of cyclin B1 and cyclin D1, which are related to cell proliferation, was not affected by TIM-3 blockade [170].

Targeting immune checkpoint molecules can revive dysfunctional γδT cells. Dysfunctional γδT cells express high levels of multiple checkpoint molecules on their surface, a phenotype similar to that of anergic or exhausted T cells. Vδ2T cells can recognize phosphoantigens with the assistance of BTN3A/BTN2A, and some subsets of Vδ1T and Vδ3T cells can recognize lipid antigens presented by CD1d and transmit activation signals through γδTCR. Activated γδT cells express NKG2D and/or other similar costimulatory molecules on their surface. Immunoglobulin-like transcripts (ILTs) or leukocyte immunoglobulin-like receptors (LIRs) belong to the Ig superfamily. ILT2 (LIRB1) binds to HLA-G in addition to recognizing other ligands and can inhibit the immune functions of γδT, NK, and B cells. NKG2A can recognize the nonclassical MHC-I molecule HLA-E and inhibit the stimulatory signal of NKG2D. Inhibitory Siglecs are immune regulatory sialic acid-binding receptors that resemble traditional immune checkpoint molecules with one or more ITIM-like motifs in the intracellular segment. Abnormal signaling of immune checkpoint molecules interferes with the normal function of TCRs, affects the level of intracellular protein phosphorylation through ITIM motifs and SHP-1/2, inhibits the proliferation and activation of γδT cells, and ultimately reduces the cytotoxicity of γδT cells. After blocking the inhibitory signals with monoclonal antibodies targeting immune checkpoint molecules, the ability of γδT cells to kill tumor cells and their interactions with other immune effector cells can be enhanced
The expression of PD-1 may be one of the reasons for the failure of Vγ9Vδ2T-cell expansion in tumor patients. Blocking the PD-1 signaling pathway can partially restore the damaged proliferation of PD-1+ γδT cells and induce their activation. A study showed that the response of bone marrow-derived Vγ9Vδ2T cells to PAgs stimulation was weakened in MM patients, while the proliferation response of γδT cells was enhanced after zoledronic acid stimulation with anti-PD-1 mAbs in vitro [51]. In addition, researchers have found that the bispecific (PD-L1× CD3) antibody Y111, which can simultaneously recognize PD-L1 and CD3, can effectively connect T cells with tumor cells expressing PD-L1. In the presence of PD-L1+ tumor cells, Y111 can induce the activation of Vγ2Vδ2T cells in a dose-dependent manner [171]. Although some studies have shown that the inhibitory effects of the PD-1 signaling pathway on γδT cells may be reversed by the synergistic effects of TCR and IL-2 signaling, blocking PD-1 signal transduction with anti-PD-L1 antibodies alone does not affect IL-2 production by γδT cells [172, 173], and the inhibitory TME also weakens TCR signal transduction. Blocking the PD-1/PD-L1 signaling pathway, restoring the proliferation and activation of γδT cells and promoting their differentiation into antitumor effector cells in the early stage of γδT cell development are important for effective ICT.
Furthermore, some studies have shown that PD-1 is not the main molecule affecting the proliferation of γδT cells and that the proliferation of γδT cells is strictly regulated by BTLA [174]. BTLA and TCR are clustered at the synapse between Vγ9Vδ2T cells and target cells, and the close localization of BTLA and TCR suggests that BTLA may affect TCR-dependent signal transduction. γδT cells need TCR signals to maintain their stability [175], and it has been suggested that the interaction between BTLA and HVEM inhibits the proliferation of Vγ9VδT cells and their response to lymphoma cells. BTLA on PB γδT cells interacted with HVEM on leukemic cells and caused some cells to stagnate in S phase; however, this did not affect the percentage of G0 cells but did increase the percentage of cells in G2/M phase. After blocking the interaction between BTLA and HVEM with mAbs, PAg/TCR-mediated signal transduction can be enhanced, and the proliferation of γδT cells can be upregulated [51, 98, 176]. However, no synergistic effect was found after the combined blockade of BTLA and PD-1, suggesting that BTLA and PD-1 may have independent effects on the proliferation and cytotoxicity of human PB γδT cells [174]. That is, inhibition of the BTLA signaling pathway can promote the proliferation of γδT cells without affecting their cytotoxicity, the production of IFN-γ or the nontargeted degranulation induced by bromohydrin pyrophosphate (BrHPP) [176].
ICIs can enhance the cytotoxicity of exhausted γδT cells
After treatment with ICIs, the cytotoxicity of γδT cells was enhanced (Fig. 5). After blocking PD-1, the antibody-dependent cell-mediated cytotoxicity (ADCC) effects of CD16+ Vγ9T cells on lymphoma cells was improved [177]. However, it has also been reported that in the environment of PD-L1+ tumor cells, blocking or knocking out PD-1 does not significantly increase the cytotoxicity of γδT cells. In contrast, anti-PD-L1 mAbs could enhance the cytotoxicity of γδT cells against some cancer cells, and the expression level of PD-L1 was positively correlated with the cytotoxicity of γδT cells [178].
Increased secretion of antitumor cytokines
After ICs are blocked, γδT cells can produce more inflammatory cytokines, especially IFN-γ and TNF-α. Inhibitory ICs may reduce the production of IFN-γ by inhibiting the key transcription factor Eomes [179]. The PD-1 signaling pathway may be involved in the regulation of IFN-γ production by γδT cells [180]. An in vitro study of γδT cells showed that, similar to that of traditional αβT cells, the cytotoxicity of activated PD1+ Vδ2T cells was inhibited, and the secretion of IFN-γ decreased after PD-L1 binding [172]. Blocking PD-1 with antibodies such as pembrolizumab can promote the secretion and release of IFN-γ and TNF-α by activated γδT cells. After the combined use of anti-LAG-3 and anti-PD-1 antibodies, the secretion of cytokines, especially IFN-γ, in γδT cells increased [92, 171, 177]. However, it should be noted that presensitization of target cells or γδT-cell activation is needed for the production of IFN-γ; that is, the regulatory effects of PD-1 signaling on the proliferation and cytokine secretion of γδT cells depends on costimulatory signals or early activation of γδT cells [92]. The TIM-3 signaling pathway also inhibits the secretion of IFN-γ and TNF-α by γδT cells. In vitro studies have shown that TIM-3+ γδT cells do not produce IFN-γ or TNF-α and have reduced cytotoxicity [96, 181]. In addition, the effects of B7-H3 on the cytokine profile of Vδ2T cells was studied. It was found that B7-H3 inhibited the expression of IFN-γ in Vδ2T cells by inhibiting T-bet [168], suggesting that the ability of γδT cells to produce cytokines would be restored after blocking TIM-3 and B7-H3 with ICIs.
The expression of effector genes in the IFN signaling pathway is negatively correlated with the degree of γδT-cell exhaustion. In exhausted T cells, several inhibitory receptors, including VSIR, KLRG1, LAG3 and TIGIT, as well as the transcription factors NR4A2 and ID2, are significantly upregulated, and IFN response genes, such as IFITM1, STAT1 and IFI6, are also upregulated [182].
Increased secretion of perforin and granzyme
After blocking ICs, the expression of perforin and granzyme is not inhibited, and cytotoxicity is enhanced. In vitro studies showed that after a PD-1 blocking drug was used and ZA was administered as stimulation, the cytotoxicity of Vγ9Vδ2T cells increased nearly 5 times, accompanied by an increase in the expression of the degranulation marker CD107 and an increase in the proportion of CD107+ Vγ9Vδ2T cells [51]. ERK1/2, STAT-3 and Wnt are known to regulate the expression of perforin and granzyme B in various immune cells. Some studies have found that increased expression of members of the TIM-3 pathway can significantly decrease the level of pERK1/2 in Vγ9Vδ2T cells activated by recombinant human (rh) Gal-9 but does not affect the level of pSTAT3 or Wnt [183]. Therefore, blocking TIM-3 can increase the killing effects of Vγ9VδT cells on colon cancer cells by activating the ERK1/2 pathway and upregulating the expression of perforin and granzyme B. B7-H3 also inhibits the cytotoxicity of Vδ2T cells by downregulating the expression of perforin and granzyme B [168], which can be reversed by using B7-H3 blockers. In tumor tissue, compared with PD-1 + LAG-3– cells, PD-1 + LAG-3+ T cells have a weaker ability to produce cytokines and/or undergo degranulation [177]. The cytotoxicity of Vδ2T cells against NSCLC tumor cell lines was enhanced after blocking PD-1 [171, 174], so the degranulation of γδT cells can be improved with the inhibition of the PD-1/PD-L1 or LAG-3 signaling pathway.
KIRs increase the threshold for Vγ9Vδ2T cell antigen-based activation, inhibiting the killing effects of cytotoxic Vγ9Vδ2T cells on MHC-I+ tumor cell lines. Blocking the binding of NKG2A to HLA-E can restore the high responsiveness of NKG2A+ Vδ2T cells. Low expression of NKG2A is usually accompanied by high expression of other ICs, such as PD-1 [141]. NKG2A is often coexpressed with PD-1, CTLA-4, LAG-3 and TIM-3 in CD8+ T cells, but they have different inhibition mechanisms [184]. The safety of humanized anti-NKG2A mAbs has been verified in clinical trials, and studies on HLA-E+ tumor cells have proven that combinations blocking the inhibitory signals of PD-1 and NKG2A have synergistic effects, which are characterized by enhanced ADCC and increased expression of CD107 and degranulation substances [108, 185].
ICIs can promote synergistic antitumor effects of γδT cells and other immune cells
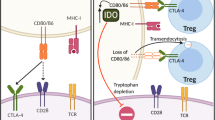
When ICLs on the surface of γδT cells are blocked, the positive interaction between γδT cells and other immune cells becomes more efficient (Fig. 5). Studies have shown that γδT cells are the key source of immunosuppressive ICLs in tumor tissues and may have the ability to regulate αβT cells. Blocking PD-L1 on γδT cells in PDAC can enhance the levels of infiltrating CD4+ and CD8+ T cells and improve immunotherapy efficacy [124]. Data from mouse models also indicate that specific γδT-cell subsets that express PD-L1 can inhibit αβT cell infiltration through PD-1/PD-L1 signaling and promote tumor growth. Vδ2T cells can also activate CTLA-4 and inhibit αβT cells through CD86. In individuals with normal γδT-cell function, the use of PD-L1 or Galectin-9 inhibitors can promote the expansion and activation of CD4+ and CD8+ T cells, but this effect is not observed in the absence of γδT cells [92, 124, 186]. In addition, PD-L1 is a downstream target of HIF1α [187]. Hypoxia and coculture with γδT cells increased the apoptosis rate of CD8+ T cells, suggesting that γδT cells can induce the death of CD8+ T cells, and this effect was significantly changed after blocking PD-1. After blocking BTNL2, the number of cytotoxic CD8+ T cells in the TME increased [126]. Monalizumab can inhibit the newly recognized IC NKG2A [188] and thus activate the antitumor effects of αβT, γδT and NK cells. Therefore, after using ICIs to reverse the immunosuppressive state of γδT cells, αβT cells can better promote tumor cell killing.
Targeting checkpoint molecules on γδT cells with ICIs can also inhibit Tregs. PD-L1 expressed by γδT cells can promote the production of Tregs and enhance the expression of the FOXP3 gene, thus maintaining the expression of TIGIT, which plays an important role in immune regulation; these effects promote Tregs to directly inhibit effector T cells through CTLA-4 and LAG-3 [71, 189]. A high TIGIT/DNAM-1 ratio was detected in Foxp3+ γδT cells of patients with AML and Tregs of patients with melanoma. High expression of TIGIT can promote the stability and inhibitory function of Tregs, which is highly correlated with poor clinical prognosis [190]. Therefore, treatment with anti-TIGIT mAbs can mediate the direct killing of tumor cells in patient tumor samples and preclinical mouse models and can also kill Tregs via Fc receptors (FCRs) [191].
In conclusion, after blocking ICR-ICL signaling with ICIs, γδT cells can gradually recover their functions and produce synergistic antitumor immune effects.
Relationship between ICI resistance and γδT cells
The rate of response to ICIs is related to many factors [192, 193], and ICT can be a good option for cancer patients who are likely to have an active immune response. However, tumor cells may evolve other mechanisms of resistance after a period of treatment [78, 194,195,196,197,198]. The mechanisms of drug resistance may not be failure of targeted drugs but rather compensatory changes in γδT cells leading to acquired resistance to drugs [72, 199]. For example, in EGFR- and KRAS-mutant mice and human lung cancer specimens, therapeutic PD-1-blocking antibodies still bind to T cells as the disease progresses; this finding suggests that mAbs still play a role when drug resistance occurs, so attention should be given to pathways other than PD-1/PD-L1 in adaptive drug resistance [200].
During the course of treatment with mAbs, compensatory and alternative suppressor receptors on γδT cells are upregulated, indicating that γδT cells may also be involved in the development of ICI tolerance in cancer patients. Some data show that anti-PD-1 inhibition significantly increases the frequency of TIM-3+ Vδ2T cells, indicating compensatory upregulation of TIM-3; additionally, combined inhibition of TIM-3 and PD-1 can significantly increase the production of TNF-α and IFN-γ [222]. For example, the upregulation of ICs does not mean the absolute exhaustion of γδT cells [141, 143]. As such, it remains to be determined how the specific epigenetic state of γδT cells changes as dysfunction develops. Furthermore, when chromatin remodeling occurs, is it reversible? How do γδT cells exhibit opposite effects under the influence of different TMEs, and can these effects be exploited? Are the changes in the expression patterns of γδT-cell ICs similar to those of αβT cells? If future research can identify a method for selective targeting of certain subsets, γδT cells will be able to play a more efficient antitumor role.
Tumor models used for the study of γδT cells urgently need to be improved for several reasons. First, the groups of γδT cells are different between humans and mice [37]. Therefore, the subgroup results obtained in mice cannot be perfectly applied to humans. Second, BTNs similar to those in humans have not been found in mice. Therefore, rodent models cannot completely represent the actual environment in humans [223]. Furthermore, the TME is different in different tumor models and is also different from that in humans.
The therapeutic prospects of targeting checkpoints on the surface of γδT cells are broad. However, resistance to ICT based on γδT can occur, which necessitates researches on the mechanism underlying resistance to γδT-cell therapy and multitarget combination therapy. Furthermore, the role of γδT cells in irAEs needs to be further clarified, which will pave the way for the inclusion of γδT cells in the arsenal of immunotherapy options.
First, MHC molecule independence and glycolipid antigen recognition activity are great advantages for γδT cells. In ICI-resistant tumor cells, a similarity between cells with ICI resistance and a hypermetabolic phenotype was observed. This phenotype included synergistic upregulation of glycolysis and mitochondrial oxidative phosphorylation, which assists tumor cells in maintaining vigorous growth under hypoxia and resisting ICT [195, 224]. The associations between checkpoint molecules related to the internal metabolism of tumor cells and ICs is also an interesting topic [225].
Second, ICs can be either costimulatory or coinhibitory molecules. Agents targeting inhibitory molecules combined with αβT cell agonists have been shown to have synergistic antitumor effects [226,227,228,229,230]. As dysfunctional γδT cells can also express multiple ICs, it may be possible to restore the antitumor potential of γδT cells. In addition, given that the mechanism of action of CD39/CD73 is relatively independent of traditional ICs and that CD39/CD73 and its related ADO pathway have been proven to be related to the survival and prognosis of patients [231], the rationality of ICI combined with other targets, such as agents targeting CD39/CD73, deserves further discussion.
In addition to considering the combination of multitarget therapy with ICT, whether combination immunotherapy based on γδT cells can have a synergistic effect is also worth exploring. Vγ9Vδ2T cells can release a large number of cytokines and chemokines to induce the activation of other bystander immune cells after being activated by amino bisphosphonic acids such as ZA. With the help of MCP-2, γδT cells can activate the effector function of granulocytes [232]. Some studies have shown that the maturation of immature DCs may mainly result from a bystander process because immature DCs rely on the activation of Vγ9Vδ2T cells by TCR signal transduction and are able to promote Vγ9Vδ2T cells to secrete cytokines required for the maturation process [233]. Bystander activation of T cells is a type of antigen-independent activation that can occur through different receptors and costimulatory signals and has been observed in cancers [234]. Microorganism-related pattern recognition receptors, such as Toll-like receptors (TLRs), and cytokines such as IL-12, IL-15, and IL-18 are associated with activating bystander T-cell responses [235]. Thus, both the innate immune properties of γδT cells and their excellent cytokine and chemokine secretion abilities may aid in the recruitment of other immune cells [236] or interactions with other bystander cells in the TME and may be involved in bystander activation. Bystander T cells rarely show an exhausted status and have excellent innate killing ability [237]; therefore, reversing the immunosuppressive state of γδT cells may allow γδT cells to promote ICI responsiveness by activating bystander cells. ICT based on γδT cells combined with strategies that target bystander T cells (such as intratumoral injection of viral peptides) may have additional beneficial effects on antitumor immunity, but this hypothesis needs to be further experimentally verified and mechanistically explored.
In clinical applications, adoptive cell infusion based on γδT cells combined with ICIs or CAR-γδT cells may be promising future research directions. The feasibility and antitumor ability of CAR-γδT cells have been preliminarily proven [238, 239], and it has been shown that CAR-γδT cells can successfully migrate into the TME and cross-present tumor-associated antigens, providing more stable and durable antitumor immunity than conventional CAR-T cells in the solid tumor environment [240,241,242]. Compared with CAR-αβT cells, CAR-γδT cells have no alloreactivity and lower incidences of off-target toxicities and cytokine release syndrome [243, 244]. Arming CAR-T cells with bacterial-derived virulence factors with strong immunomodulatory properties can mediate bystander immunity through epitope spreading and can expand the therapeutic spectrum [245]. Antigen recognition by γδT cells is closely related to microbial metabolites, indicating that CAR-γδT cells may benefit from microbial engineering. CAR-γδT cells have just taken the first step toward clinical application [246, 247], and more clinical trials are required to determine the potential of CAR-γδT cells. In clinical trials, excellent antitumor activity and good patient tolerance were demonstrated for adoptive γδT-cell-based therapies after γδT cells were expanded in the presence of appropriate doses of ZA or BrHPP or infused with IL-2/IL-21 [248]. One study showed that Vδ2T cells were able to maintain a low PD-1 expression state in vivo after administration of immunotherapy [249]. However, not all patients respond to adoptive Vγ9Vδ2T-cell therapies. Considering that γδT cells present in the suppressive TME can display exhaustion or anergy, combination with ICIs might improve the cytotoxicity of γδT-cell immunotherapy and lead to better antitumor effects [250].
First describing the interaction between the TME and γδT cells, this paper has summarized the potential of γδT cells to participate in ICT by describing the functional regulation of ICs on γδT cells. Agents targeting ICs can significantly regulate the proliferation, activation and cytotoxicity of γδT cells, which provides a strategy to reverse the immunosuppressive state of γδT cells and supports the use of ICT based on γδT cells in clinical applications in the future.
Availability of data and materials
Not applicable.
Abbreviations
- Abt:
-
Antibiotic-treated
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ADO:
-
Adenosine
- AML:
-
Acute myelogenous leukemia
- AMP:
-
Adenosine monophosphate
- A2AR:
-
A2A receptor
- BrHPP:
-
Bromohydrin pyrophosphate
- BTLA:
-
B- and T-lymphocyte attenuator
- BTN2A:
-
Butyrophilin 2A
- BTN3A:
-
Butyrophilin 3A
- CAR:
-
Chimeric antigen receptor
- CCL2:
-
C-C motif chemokine 2
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DNT:
-
Double-negative T cell
- eATP:
-
Extracellular adenosine triphosphate
- FCR:
-
Fc receptor
- GBM:
-
Glioblastoma
- GC:
-
Germinal center
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HIF:
-
Hypoxia-inducible factor
- HMBPP:
-
(e)-4-hydroxy-3-methyl-but-2-enylpyrophosphate
- HMGB-1:
-
High mobility group box protein 1
- IC:
-
Immune checkpoint
- ICB:
-
Immune checkpoint blockade
- ICI:
-
Immune checkpoint inhibitor
- ICL:
-
Immune checkpoint ligand
- ICR:
-
Immune checkpoint receptor
- IFN-γ:
-
Interferon-gamma
- IL:
-
Interleukin
- ILT2:
-
Ig-like transcript 2
- IPP:
-
Isopentenyl pyrophosphate
- ITIM:
-
Immune receptor tyrosine-based inhibition motif
- irAEs:
-
Immune-related adverse events
- ITSM:
-
Immune receptor tyrosine-based switch motif
- LAG-3:
-
Lymphocyte-activation gene 3
- MCP-1:
-
Monocyte chemoattractant protein 1
- MCP-2:
-
Monocyte chemoattractant protein 2
- MHC:
-
Major histocompatibility complex
- MM:
-
Multiple myeloma
- MDSC:
-
Myeloid-derived suppressor cell
- OS:
-
Overall survival
- TAM:
-
Tumor-associated macrophage
- TCR:
-
T-cell receptor
- TGF:
-
Transforming growth factor-beta
- TIM-3:
-
T-cell immunoglobulin domain and mucin domain-3
- TIL:
-
Tumor-infiltrating lymphocyte
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TMG:
-
Tumor marker ganglioside
- TNF-α:
-
Tumor necrosis factor-alpha
- PAg:
-
Phosphoantigen
- PB:
-
Peripheral blood
- PDAC:
-
Pancreatic ductal adenocarcinoma
- pDC:
-
Plasmacytoid dendritic cell
- pMHC:
-
Peptide-MHC-I complexes
- ROS:
-
Reactive oxygen species
- SHM:
-
Somatic hypermutation
- SHP:
-
Scr homology region 2 domain-containing phosphatase
- Siglec:
-
Sialic acid binding immunoglobulin-like lectin
- STAT:
-
Signal transducer and activator of transcription
- ZA:
-
Zoledronic acid
References
Zakeri N, Hall A, Swadling L, Pallett LJ, Schmidt NM, Diniz MO, et al. Characterisation and induction of tissue-resident gamma delta T-cells to target hepatocellular carcinoma. Nat Commun. 2022;13:1372.
Wu Y, Biswas D, Usaite I, Angelova M, Boeing S, Karasaki T, et al. A local human Vδ1 T cell population is associated with survival in nonsmall-cell lung cancer. Nat Cancer. 2022;3:696–709.
Mikulak J, Oriolo F, Bruni E, Roberto A, Colombo FS, Villa A, et al. NKp46-expressing human gut-resident intraepithelial Vδ1 T cell subpopulation exhibits high antitumor activity against colorectal cancer. JCI Insight. 2019;4:125884.
He W, Hu Y, Chen D, Li Y, Ye D, Zhao Q, et al. Hepatocellular carcinoma-infiltrating γδ T cells are functionally defected and allogenic Vδ2+ γδ T cell can be a promising complement. Clin Transl Med. 2022;12:e800 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ctm2.800. Cited 2022 Sep 13.
Bruni E, Cimino MM, Donadon M, Carriero R, Terzoli S, Piazza R, et al. Intrahepatic CD69+Vδ1 T cells re-circulate in the blood of patients with metastatic colorectal cancer and limit tumor progression. J Immunother Cancer. 2022;10:e004579.
Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. 2009;31:184–96.
Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13:88–100.
Silva-Santos B, Serre K, Norell H. γδ T cells in cancer. Nat Rev Immunol. 2015;15:683–91.
Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19:169–84.
Conejo-Garcia JR, Innamarato P. γδ T cells share the spotlight in cancer. Nat Cancer. 2022;3:657–8.
Xu Y, **ang Z, Alnaggar M, Kouakanou L, Li J, He J, et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell Mol Immunol. 2021;18:427–39.
Natasja L, de Vries Joris, van de Haar Vivien, Veninga Myriam, Chalabi Marieke E, Ijsselsteijn Manon, et al. Voest γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature. https://doi.org/10.1038/s41586-022-05593-1.
Saura-Esteller J, de Jong M, King LA, Ensing E, Winograd B, de Gruijl TD, et al. Gamma Delta T-Cell based Cancer immunotherapy: past-present-future. Front Immunol. 2022;13:915837 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9245381/. Cited 2022 Oct 3.
Jaeger N, Colonna M. A γδ T–cell Imprint in a Rare Skin Tumor. Cancer Immunol Res. 2021;9:600. https://doi.org/10.1158/2326-6066.CIR-21-0270 Available from: Cited 2022 May 10.
Park JH, Kim H-J, Kim CW, Kim HC, Jung Y, Lee H-S, et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat Immunol. 2021;22:336–46.
Chabab G, Barjon C, Bonnefoy N, Lafont V. Pro-tumor γδ T cells in human Cancer: polarization, mechanisms of action, and implications for therapy. Front Immunol. 2020;11:2186.
Andreu-Ballester JC, Galindo-Regal L, Hidalgo-Coloma J, Cuéllar C, García-Ballesteros C, Hurtado C, et al. Differences in circulating γδ T cells in patients with primary colon cancer and relation with prognostic factors. PLoS One. 2020;15:e0243545.
Janssen A, Villacorta Hidalgo J, Beringer DX, van Dooremalen S, Fernando F, van Diest E, et al. γδ T-cell receptors derived from breast Cancer-infiltrating T lymphocytes mediate antitumor reactivity. Cancer Immunol Res. 2020;8:530–43.
Ou L, Wang H, Huang H, Zhou Z, Lin Q, Guo Y, et al. Preclinical platforms to study therapeutic efficacy of human γδ T cells. Clin Transl Med. 2022;12:e814.
Teng MWL, Galon J, Fridman W-H, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015;125:3338–46.
Nakamura K, Smyth MJ, Martinet L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood. 2020;136:2731–40.
Gupta RG, Li F, Roszik J, Lizée G. Exploiting tumor neoantigens to target cancer evolution: current challenges and promising therapeutic approaches. Cancer Discov. 2021;11:1024–39 Available from: https://aacrjournals.org/cancerdiscovery/article/11/5/1024/666345/Exploiting-Tumor-Neoantigens-to-Target-Cancer. Cited 2022 Oct 3.
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93.
Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298–312.
Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–9.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50 Nature Publishing Group. Available from: https://www.nature.com/articles/s41591-018-0014-x. Cited 2022 Aug 25.
Bagchi S, Yuan R, Engleman EG. Immune Checkpoint inhibitors for the treatment of Cancer: clinical impact and mechanisms of response and Resistance. Annu Rev Pathol. 2021;16:223–49.
de Miguel M, Calvo E. Clinical challenges of Immune Checkpoint inhibitors. Cancer Cell. 2020;38:326–33.
Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discov. 2021;20:899–919 Nature Publishing Group. Available from: https://www.nature.com/articles/s41573-021-00155-y. Cited 2022 Aug 21.
Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309–37 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867421011016. Cited 2022 May 31.
Brauneck F, Weimer P, Schulze zur Wiesch J, Weisel K, Leypoldt L, Vohwinkel G, et al. Bone marrow-resident Vδ1 T cells co-express TIGIT with PD-1, TIM-3 or CD39 in AML and myeloma. Front Med-Lausanne. 2021;8 Available from: https://www.frontiersin.org/articles/10.3389/fmed.2021.763773. Cited 2022 Aug 20.
Haas W, Pereira P, Tonegawa S. Gamma/delta cells. Annu Rev Immunol. 1993;11:637–85.
Reis BS, Darcy PW, Khan IZ, Moon CS, Kornberg AE, Schneider VS, et al. TCR-Vγδ usage distinguishes protumor from antitumor intestinal γδ T cell subsets. Science. 2022;377:276–84.
Lafont V, Sanchez F, Laprevotte E, Michaud H-A, Gros L, Eliaou J-F, et al. Plasticity of gamma delta T cells: impact on the anti-tumor response. Front Immunol. 2014;5 Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2014.00622. Cited 2022 Aug 2.
Bottino C, Tambussi G, Ferrini S, Ciccone E, Varese P, Mingari MC, et al. Two subsets of human T lymphocytes expressing gamma/delta antigen receptor are identifiable by monoclonal antibodies directed to two distinct molecular forms of the receptor. J Exp Med. 1988;168:491–505.
Dang W, Pin W, Qiu F, Wei Q, Huang J. Human γδT-cell subsets and their involvement in tumor immunity. Cell Mol Immunol. 2017;14:245–53.
O’Brien RL, Born WK. Two functionally distinct subsets of IL-17 producing γδ T cells. Immunol Rev. 2020;298:10–24.
Casetti R, Agrati C, Wallace M, Sacchi A, Martini F, Martino A, et al. Cutting edge: TGF-beta1 and IL-15 induce FOXP3+ gammadelta regulatory T cells in the presence of antigen stimulation. J Immunol. 2009;183:3574–7.
Peters C, Meyer A, Kouakanou L, Feder J, Schricker T, Lettau M, et al. TGF-β enhances the cytotoxic activity of Vδ2 T cells. Oncoimmunology. 2019;8:e1522471.
Lo Presti E, Toia F, Oieni S, Buccheri S, Turdo A, Mangiapane LR, et al. Squamous Cell tumors recruit γδ T cells producing either IL17 or IFNγ depending on the Tumor stage. Cancer Immunol Res. 2017;5:397–407.
Ryan PL, Sumaria N, Holland CJ, Bradford CM, Izotova N, Capucine L, et al. Heterogeneous yet stable Vδ2(+) T-cell profiles define distinct cytotoxic effector potentials in healthy human individuals. Proc Natl Acad Sci. 2016;113:14378–83 Available from: https://www.pnas.org/doi/full/10.1073/pnas.1611098113. Cited 2022 Jul 17.
Paquin-Proulx D, Barsotti NS, Santos BAN, Ana KB, Marinho B, Kokron CM, et al. Inversion of the Vδ1 to Vδ2 γδ T cell ratio in CVID is not restored by IVIg and is associated with immune activation and exhaustion. Medicine. 2016;95:e4304 Available from: https://journals.lww.com/md-journal/Fulltext/2016/07260/Inversion_of_the_V_1_to_V_2____T_cell_ratio_in.42.aspx. Cited 2022 Jul 17.
Lo Presti E, Dieli F, Fourniè JJ, Meraviglia S. Deciphering human γδ T cell response in cancer: lessons from tumor-infiltrating γδ T cells. Immunol Rev. 2020;298:153–64.
Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med. 2019;11:eaax9364.
Lu H, Dai W, Guo J, Wang D, Wen S, Yang L, et al. High abundance of Intratumoral γδ T cells favors a better prognosis in head and neck squamous Cell carcinoma: a Bioinformatic analysis. Front Immunol. 2020;11:573920.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Lee M, Park C, Woo J, Kim J, Kho I, Nam D-H, et al. Preferential infiltration of unique Vγ9Jγ2-Vδ2 T cells into glioblastoma Multiforme. Front Immunol. 2019;10:555.
Bryant NL, Gillespie GY, Lopez RD, Markert JM, Cloud GA, Langford CP, et al. Preclinical evaluation of ex vivo expanded/activated γδ T cells for immunotherapy of glioblastoma multiforme. J Neuro-Oncol. 2011;101:179–88.
Chan KF, Duarte JDG, Ostrouska S, Behren A. γδ T cells in the Tumor Microenvironment-interactions with other Immune cells. Front Immunol. 2022;13:894315.
Girard P, Ponsard B, Charles J, Chaperot L, Aspord C. Potent bidirectional cross-talk between Plasmacytoid dendritic cells and γδT cells through BTN3A, type I/II IFNs and Immune checkpoints. Front Immunol. 2020;11:861.
Castella B, Foglietta M, Sciancalepore P, Rigoni M, Coscia M, Griggio V, et al. Anergic bone marrow Vγ9Vδ2 T cells as early and long-lasting markers of PD-1-targetable microenvironment-induced immune suppression in human myeloma. Oncoimmunology. 2015;4:e1047580.
Petrasca A, Melo AM, Breen EP, Doherty DG. Human Vδ3+ γδ T cells induce maturation and IgM secretion by B cells. Immunol Lett. 2018;196:126–34 Available from: https://www.sciencedirect.com/science/article/pii/S0165247817304443. Cited 2022 Aug 20.
Horner AA, Jabara H, Ramesh N, Geha RS. Gamma/delta T lymphocytes express CD40 ligand and induce isotype switching in B lymphocytes. J Exp Med. 1995;181:1239–44.
Caccamo N, Battistini L, Bonneville M, Poccia F, Fournié JJ, Meraviglia S, et al. CXCR5 Identifies a Subset of Vγ9Vδ2 T Cells which Secrete IL-4 and IL-10 and Help B Cells for Antibody Production. J Immunol. 2006;177:5290–5 Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.177.8.5290. Cited 2022 Jul 16.
Ullrich L, Lueder Y, Juergens A-L, Wilharm A, Barros-Martins J, Bubke A, et al. IL-4-producing Vγ1+/Vδ6+ γδ T cells sustain germinal center reactions in Peyer’s patches of mice. Front Immunol. 2021;12:729607.
Yang Y, Li L, Yuan L, Zhou X, Duan J, **ao H, et al. A structural change in Butyrophilin upon Phosphoantigen binding underlies Phosphoantigen-Mediated Vγ9Vδ2 T Cell activation. Immunity. 2019;50:1043–1053.e5.
Siyi G, Borowska MT, Boughter CT, Adams EJ. Butyrophilin3A proteins and Vγ9Vδ2 T cell activation. Semin Cell Dev Biol. 2018;84:65–74.
Morita CT, Beckman EM, Bukowski JF, Tanaka Y, Band H, Bloom BR, et al. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity. 1995;3:495–507.
Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat Immunol. 2013;14:908–16 Nature Publishing Group. Available from: https://www.nature.com/articles/ni.2665. Cited 2022 Sep 26.
Riaño F, Karunakaran MM, Starick L, Li J, Scholz CJ, Kunzmann V, et al. Vγ9Vδ2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome. Eur J Immunol. 2014;44:2571–6 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/eji.201444712. Cited 2022 Sep 26.
Djaoud Z, Parham P. HLAs, TCRs, and KIRs, a triumvirate of human Cell-Mediated Immunity. Annu Rev Biochem. 2020;89:717–39.
Boutin L, Scotet E. Towards deciphering the hidden mechanisms that contribute to the antigenic activation process of human Vγ9Vδ2 T cells. Front Immunol. 2018;9:828.
Rigau M, Uldrich AP, Behren A. Targeting butyrophilins for cancer immunotherapy. Trends Immunol. 2021;42:670–80.
Sandstrom A, Peigné C-M, Léger A, Crooks JE, Konczak F, Gesnel M-C, et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity. 2014;40:490–500.
Benyamine A, Loncle C, Foucher E, Blazquez J-L, Castanier C, Chrétien A-S, et al. BTN3A is a prognosis marker and a promising target for Vγ9Vδ2 T cells based-immunotherapy in pancreatic ductal adenocarcinoma (PDAC). Oncoimmunology. 2017;7:e1372080.
Vantourout P, Laing A, Woodward MJ, Zlatareva I, Apolonia L, Andrew W, et al. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc Natl Acad Sci. 2018;115:1039–44 Available from: https://www.pnas.org/doi/full/10.1073/pnas.1701237115. Cited 2022 Sep 1.
Rhodes DA, Chen H-C, Williamson JC, Hill A, Yuan J, Smith S, et al. Regulation of human γδ T cells by BTN3A1 protein stability and ATP-binding cassette transporters. Front Immunol. 2018;9:662.
Riganti C, Castella B, Massaia M. ABCA1, apoA-I, and BTN3A1: a legitimate Ménage à trois in dendritic cells. Front Immunol. 2018;9:1246.
Laplagne C, Ligat L, Foote J, Lopez F, Fournié J-J, Laurent C, et al. Self-activation of Vγ9Vδ2 T cells by exogenous phosphoantigens involves TCR and butyrophilins. Cell Mol Immunol. 2021;18:1861–70.
Castella B, Riganti C, Massaia M. Metabolic approaches to rescue antitumor Vγ9Vδ2 T-cell functions in myeloma. Front Biosci-Landmrk. 2020;25:69–105 IMR Press. Available from: https://www.imrpress.com/journal/FBL/25/1/10.2741/4795. Cited 2022 Jul 16.
Miyashita M, Shimizu T, Ashihara E, Ukimura O. Strategies to improve the antitumor effect of γδ T Cell immunotherapy for clinical application. Int J Mol Sci. 2021;22:8910.
Wesch D, Kabelitz D. Hans-Heinrich Oberg. Tumor resistance mechanisms and their consequences on γδ T cell activation. Immunol Rev. 2020;298:84–98 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/imr.12925. Cited 2022 May 31.
Mao Y, Yin S, Zhang J, Hu Y, Huang B, Cui L, et al. A new effect of IL-4 on human γδ T cells: promoting regulatory Vδ1 T cells via IL-10 production and inhibiting function of Vδ2 T cells. Cell Mol Immunol. 2016;13:217–28.
Peters C, Kabelitz D. Daniela Wesch. Regulatory functions of γδ T cells. Cell Mol Life Sci. 2018;75:2125–35 Available from: http://springer.longhoe.net/10.1007/s00018-018-2788-x. Cited 2022 Jul 16.
Sabbione F, Gabelloni ML, Ernst G, Gori MS, Salamone G, Oleastro M, et al. Neutrophils suppress γδ T-cell function. Eur J Immunol. 2014;44:819–30.
Kersten K, Coffelt SB, Hoogstraat M, Verstegen NJM, Vrijland K, Ciampricotti M, et al. Mammary tumor-derived CCL2 enhances pro-metastatic systemic inflammation through upregulation of IL1β in tumor-associated macrophages. Oncoimmunology. 2017;6:e1334744.
Zuberbuehler MK, Parker ME, Wheaton JD, Espinosa JR, Salzler HR, Park E, et al. The transcription factor c-Maf is essential for the commitment of IL-17-producing γδ T cells. Nat Immunol. 2019;20:73–85.
Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, et al. CD73 expression on effector T cells sustained by TGF-β facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat Commun. 2019;10:150.
Zhao H, Bo C, Kang Y, Li H. What Else can CD39 tell us? Front Immunol. 2017;8:727.
Otsuka A, Hanakawa S, Miyachi Y, Kabashima K. CD39: a new surface marker of mouse regulatory γδ T cells. J Allergy Clin Immunol. 2013;132:1448–51.
Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–9 Available from: https://www.nature.com/articles/s41591-019-0522-3. Nature Publishing Group; Cited 2022 Sep 18.
Deng W-W, Li Y-C, Ma S-R, Mao L, Yu G-T, Bu L-L, et al. Specific blockade CD73 alters the “exhausted” phenotype of T cells in head and neck squamous cell carcinoma. Int J Cancer. 2018;143:1494–504.
Gallerano D, Ciminati S, Grimaldi A, Piconese S, Cammarata I, Focaccetti C, et al. Genetically driven CD39 expression shapes human tumor-infiltrating CD8+ T-cell functions. Int J Cancer. 2020;147:2597–610.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–8.
** C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell. 2019;176:998–1013.e16.
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40:785–800 Elsevier; Cited 2022 Jul 16. Available from: https://www.cell.com/immunity/abstract/S1074-7613(14)00147-2.
Gruenbacher G, Gander H, Rahm A, Idzko M, Nussbaumer O, Thurnher M. Ecto-ATPase CD39 inactivates isoprenoid-derived Vγ9Vδ2 T Cell Phosphoantigens. Cell Rep. 2016;16:444–56.
Jandke A, Melandri D, Monin L, Ushakov DS, Laing AG, Vantourout P, et al. Butyrophilin-like proteins display combinatorial diversity in selecting and maintaining signature intraepithelial γδ T cell compartments. Nat Commun. 2020;11:3769.
Willcox CR, Vantourout P, Salim M, Zlatareva I, Melandri D, Zanardo L, et al. Butyrophilin-like 3 directly binds a human Vγ4+ T Cell receptor using a modality distinct from clonally-restricted antigen. Immunity. 2019;51:813–825.e4.
Castella B, Melaccio A, Foglietta M, Riganti C, Massaia M. Vγ9Vδ2 T cells as strategic weapons to improve the potency of Immune Checkpoint blockade and Immune interventions in human myeloma. Front Oncol. 2018;8:508.
Hsu H, Boudova S, Mvula G, Divala TH, Mungwira RG, Harman C, et al. Prolonged PD1 expression on neonatal Vδ2 lymphocytes dampens Proinflammatory responses: role of epigenetic regulation. J Immunol. 2016;197:1884–92.
Hoeres T, Holzmann E, Smetak M, Birkmann J, Wilhelm M. PD-1 signaling modulates interferon-γ production by Gamma Delta (γδ) T-Cells in response to leukemia. OncoImmunology. 2019;8:1550618. Taylor & Francis; Available from: Cited 2022 May 10. https://doi.org/10.1080/2162402X.2018.1550618.
Yi H, Chen D, Hong M, Liu J, Li Y, Hao J, et al. Apoptosis, Pyroptosis, and Ferroptosis conspiringly induce immunosuppressive hepatocellular carcinoma Microenvironment and γδ T-Cell imbalance. Front Immunol. 2022;13:845974.
Macek JZ. Gamma delta T cells in hepatocellular carcinoma: sunrise of new therapy based on Vδ2 T cells? Clin Transl Med. 2022;12:e834.
Gherardin NA, Waldeck K, Caneborg A, Martelotto LG, Balachander S, Zethoven M, et al. γδ T cells in merkel cell carcinomas have a proinflammatory profile prognostic of patient survival. Cancer Immunol Res. 2021;9:612–23. Available from. Cited 2022 Jun 3. https://doi.org/10.1158/2326-6066.CIR-20-0817.
** Z, Lan T, Zhao Y, **xia D, Chen J, Lai J, et al. Higher TIGIT+CD226- γδ T cells in patients with acute myeloid leukemia. Immunol Investig. 2022;51:40–50.
Wu K, Feng J, **u Y, Li Z, Lin Z, Zhao H, et al. Vδ2 T cell subsets, defined by PD-1 and TIM-3 expression, present varied cytokine responses in acute myeloid leukemia patients. Int Immunopharmacol. 2020;80:106122.
Catafal-Tardos E, Baglioni MV, Bekiaris V. Inhibiting the unconventionals: importance of immune checkpoint receptors in γδ T, MAIT, and NKT cells. Cancers. 2021;13:4647.
Gorgulho J, Roderburg C, Heymann F, Schulze-Hagen M, Beier F, Vucur M, et al. Serum levels of soluble B and T lymphocyte attenuator predict overall survival in patients undergoing immune checkpoint inhibitor therapy for solid malignancies. Int J Cancer. 2021;149:1189–98.
Zhan Y, Zheng L, Liu J, Dongzhi H, Wang J, Liu K, et al. PLA2G4A promotes right-sided colorectal cancer progression by inducing CD39+γδ Treg polarization. JCI Insight. 2021;6:148028.
Girard P, Charles J, Cluzel C, Degeorges E, Manches O, Plumas J, et al. The features of circulating and tumor-infiltrating γδ T cells in melanoma patients display critical perturbations with prognostic impact on clinical outcome. OncoImmunology. 2019;8:1601483. Available from: Taylor & Francis. Cited 2022 May 14. https://doi.org/10.1080/2162402X.2019.1601483.
López-Botet M, Bellón T, Llano M, Navarro F, García P, de Miguel M. Paired inhibitory and triggering NK cell receptors for HLA class I molecules. Hum Immunol. 2000;61:7–17.
Crespí C. Skewed inhibitory receptors expression in a TAP2-deficient patient. Immunol Lett. 2003;86:149–53 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0165247803000051. Cited 2022 Sep 17.
Trichet V, Benezech C, Dousset C, Gesnel M-C, Bonneville M, Breathnach R. Complex interplay of activating and inhibitory signals received by Vgamma9Vdelta2 T cells revealed by target cell beta2-microglobulin knockdown. J Immunol. 2006;177:6129–36.
Harly C, Peyrat M-A, Netzer S, Déchanet-Merville J, Bonneville M, Scotet E. Up-regulation of cytolytic functions of human Vδ2-γ T lymphocytes through engagement of ILT2 expressed by tumor target cells. Blood. 2011;117:2864–73.
Lesport E, Baudhuin J, Sousa S, LeMaoult J, Zamborlini A, Rouas-Freiss N, et al. Inhibition of human gamma delta [corrected] T-cell antitumoral activity through HLA-G: implications for immunotherapy of cancer. Cell Mol Life Sci. 2011;68:3385–99.
Mingari MC, Pietra G, Moretta L. Immune Checkpoint Inhibitors: Anti-NKG2A Antibodies on Board. Trends Immunol. 2019;40:83–5 Available from: https://www.sciencedirect.com/science/article/pii/S1471490618302485. Cited 2022 Aug 24.
Borst L, van der Burg SH, van Hall T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin Cancer Res. 2020;26:5549–56. Available from: Cited 2022 Aug 24. https://doi.org/10.1158/1078-0432.CCR-19-2095.
Angelini DF, Zambello R, Galandrini R, Diamantini A, Placido R, Micucci F, et al. NKG2A inhibits NKG2C effector functions of γδ T cells: implications in health and disease. J Leukoc Biol. 2011;89:75–84.
Cazzetta V, Bruni E, Terzoli S, Carenza C, Franzese S, Piazza R, et al. NKG2A expression identifies a subset of human Vδ2 T cells exerting the highest antitumor effector functions. Cell Rep. 2021;37:109871.
Borst L, Sluijter M, Sturm G, Charoentong P, Santegoets SJ, van Gulijk M, et al. NKG2A is a late immune checkpoint on CD8 T cells and marks repeated stimulation and cell division. Int J Cancer. 2022;150:688–704.
Magalhães A, Duarte HO, Reis CA. Aberrant glycosylation in Cancer: a Novel molecular mechanism controlling metastasis. Cancer Cell. 2017;31:733–5.
Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55.
Zhang Z, Yang C, Li L, Zhu Y, Su K, Zhai L, et al. “γδT Cell-IL17A-neutrophil” Axis drives immunosuppression and confers breast Cancer Resistance to high-dose anti-VEGFR2 therapy. Front Immunol. 2021;12:699478.
van de Wall S, Santegoets KCM, van Houtum EJH, Büll C, Adema GJ. Sialoglycans and Siglecs can shape the Tumor Immune Microenvironment. Trends Immunol. 2020;41:274–85.
Bartish M, del Rincón SV, Rudd CE, Uri Saragovi H. Aiming for the sweet spot: Glyco-Immune checkpoints and γδ T cells in targeted immunotherapy. Front Immunol. 2020;11 Available from: https://www.frontiersin.org/article/10.3389/fimmu.2020.564499. Cited 2022 Jun 3.
Cagnoni AJ, Giribaldi ML, Blidner AG, Cutine AM, Gatto SG, Morales RM, et al. Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8+ regulatory T cells. P Natl Acad Sci USA. 2021;118:e2102950118.
Yang R, Sun L, Li C-F, Wang Y-H, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12:832.
Manni M, Läubli H. Targeting glyco-immune checkpoints for cancer therapy. Expert Opin Biol Th. 2021;21:1063–71. Available from: Taylor & Francis. Cited 2022 Aug 21. https://doi.org/10.1080/14712598.2021.1882989.
Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou T-D, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. 2016;17:1273–81 Nature Publishing Group. Available from: https://www.nature.com/articles/ni.3552. Cited 2022 Aug 25.
Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, et al. Sialic acid blockade suppresses tumor growth by enhancing T-cell–mediated tumor immunity. Cancer Res. 2018;78:3574–88. Available from: Cited 2022 Aug 25. https://doi.org/10.1158/0008-5472.CAN-17-3376.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9.
Garnier J, Turrini O, Chretien A-S, Olive D. Local ablative therapy associated with immunotherapy in locally advanced pancreatic cancer: a solution to overcome the double trouble?-a comprehensive review. J Clin Med. 2022;11:1948.
Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. γδ T cells support pancreatic oncogenesis by restraining αβ T Cell activation. Cell. 2016;166:1485–1499.e15.
Lopes N, Silva-Santos B. Functional and metabolic dichotomy of murine γδ T cell subsets in cancer immunity. Eur J Immunol. 2021;51:17–26.
Yanyun D, Qianwen Peng D, Cheng TP, Sun W, Wang H, et al. Cancer cell-expressed BTNL2 facilitates tumour immune escape via engagement with IL-17A-producing γδ T cells. Nat Commun. 2022;13:231.
Vitiello GA, Miller G. Targeting the interleukin-17 immune axis for cancer immunotherapy. J Exp Med. 2020;217:e20190456.
Zhang H, Chai W, Yang W, Han W, Mou W, ** Y, et al. The increased IL-17-producing γδT cells promote tumor cell proliferation and migration in neuroblastoma. Clin Immunol. 2020;211:108343 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1521661619303857. Cited 2022 Sep 7.
Patil RS, Shah SU, Shrikhande SV, Goel M, Dikshit RP, Chiplunkar SV. IL17 producing γδT cells induce angiogenesis and are associated with poor survival in gallbladder cancer patients. Int J Cancer. 2016;139:869–81 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ijc.30134. Cited 2022 Aug 14.
Wakita D, Sumida K, Iwakura Y, Nishikawa H, Ohkuri T, Chamoto K, et al. Tumor-infiltrating IL-17-producing γδ T cells support the progression of tumor by promoting angiogenesis. Eur J Immunol. 2010;40:1927–37 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/eji.200940157. Cited 2022 Aug 2.
Aotsuka A, Matsumoto Y, Arimoto T, Kawata A, Ogishima J, Taguchi A, et al. Interleukin-17 is associated with expression of programmed cell death 1 ligand 1 in ovarian carcinoma. Cancer Sci. 2019;110:3068–78.
Moesta AK, Li X-Y, Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol. 2020;20:739–55 Available from: http://www.nature.com/articles/s41577-020-0376-4. Cited 2022 Sep 20.
Zeng J, Ning Z, Wang Y, **ong H. Implications of CD39 in immune-related diseases. Int Immunopharmacol. 2020;89:107055.
Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and anti-CTLA-4 in preclinical models. Cancer Immunol Res. 2018;6:1136–49.
Chabab G, Barjon C, Abdellaoui N, Salvador-Prince L, Dejou C, Michaud H-A, et al. Identification of a regulatory Vδ1 gamma delta T cell subpopulation expressing CD73 in human breast cancer. J Leukoc Biol. 2020;107:1057–67.
Ni C, Fang Q-Q, Chen W-Z, Jiang J-X, Jiang Z, Ye J, et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+γδ1 Treg cells. Signal Transduct Tar. 2020;5:41.
Guoming H, Pin W, Cheng P, Zhang Z, Wang Z, **uyan Y, et al. Tumor-infiltrating CD39+γδTregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology. 2017;6:e1277305.
Weimer P, Wellbrock J, Sturmheit T, Oliveira-Ferrer L, Ding Y, Menzel S, et al. Tissue-specific expression of TIGIT, PD-1, TIM-3, and CD39 by γδ T cells in ovarian Cancer. Cells-Basel. 2022;11:964.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9 Nature Publishing Group. Available from: https://www.nature.com/articles/ni.2035. Cited 2022 Jul 16.
Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1471490613001543Cited 2022 Aug 18.
Blank CU, Nicholas Haining W, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19:665–74 Available from: http://www.nature.com/articles/s41577-019-0221-9. Cited 2022 Aug 18.
John Wherry E, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99 Available from: http://www.nature.com/articles/nri3862. Cited 2022 Aug 18.
ElTanbouly MA, Noelle RJ. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat Rev Immunol. 2021;21:257–67 Nature Publishing Group. Available from: https://www.nature.com/articles/s41577-020-00454-2. Cited 2022 Aug 25.
Wang Y, Zhao N, Zhang X, Li Z, Liang Z, Yang J, et al. Bibliometrics analysis of Butyrophilins as Immune regulators [1992-2019] and implications for Cancer prognosis. Front Immunol. 2020;11:1187.
He D, Qin Z, Liu Z, Ji X, Gao J, Guo H, et al. Comprehensive analysis of the prognostic value and Immune infiltration of Butyrophilin subfamily 2/3 (BTN2/3) members in Pan-glioma. Front Oncol. 2022;12:816760.
Blazquez J-L, Benyamine A, Pasero C, Olive D. New insights into the regulation of γδ T cells by BTN3A and other BTN/BTNL in Tumor Immunity. Front Immunol. 2018;9:1601.
Bian B, Fanale D, Dusetti N, Roque J, Pastor S, Chretien A-S, et al. Prognostic significance of circulating PD-1, PD-L1, pan-BTN3As, BTN3A1 and BTLA in patients with pancreatic adenocarcinoma. Oncoimmunology. 2019;8:e1561120.
Cai P, Lu Z, Wu J, Qin X, Wang Z, Zhang Z, et al. BTN3A2 serves as a prognostic marker and favors immune infiltration in triple-negative breast cancer. J Cell Biochem. 2020;121:2643–54.
Incorvaia L, Fanale D, Badalamenti G, Porta C, Olive D, De Luca I, et al. Baseline plasma levels of soluble PD-1, PD-L1, and BTN3A1 predict response to nivolumab treatment in patients with metastatic renal cell carcinoma: a step toward a biomarker for therapeutic decisions. Oncoimmunology. 2020;9:1832348.
Billon E, Chanez B, Rochigneux P, Albiges L, Vicier C, Pignot G, et al. Soluble BTN2A1 Is a potential prognosis biomarker in pre-treated advanced renal Cell carcinoma. Front Immunol. 2021;12:670827.
Fanale D, Incorvaia L, Badalamenti G, De Luca I, Algeri L, Bonasera A, et al. Prognostic role of plasma PD-1, PD-L1, pan-BTN3As and BTN3A1 in patients affected by metastatic gastrointestinal stromal tumors: can Immune checkpoints act as a sentinel for short-term survival? Cancers. 2021;13:2118.
Benyamine A, Le Roy A, Mamessier E, Gertner-Dardenne J, Castanier C, Orlanducci F, et al. BTN3A molecules considerably improve Vγ9Vδ2T cells-based immunotherapy in acute myeloid leukemia. Oncoimmunology. 2016;5:e1146843 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5087298/. Cited 2022 Sep 6.
Di Marco BR, Roberts NA, Dart RJ, Vantourout P, Jandke A, Nussbaumer O, et al. Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific γδ T Cell Compartments. Cell. 2016;167:203–218.e17 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867416310819. Cited 2022 Sep 17.
Harly C, Guillaume Y, Nedellec S, Peigné C-M, Mönkkönen H, Mönkkönen J, et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood. 2012;120:2269–79.
Starick L, Riano F, Karunakaran MM, Kunzmann V, Li J, Kreiss M, et al. Butyrophilin 3A (BTN3A, CD277)-specific antibody 20.1 differentially activates Vγ9Vδ2 TCR clonotypes and interferes with phosphoantigen activation. Eur J Immunol. 2017;47:982–92.
De Gassart A, Le K-S, Brune P, Agaugué S, Sims J, Goubard A, et al. Development of ICT01, a first-in-class, anti-BTN3A antibody for activating Vγ9Vδ2 T cell-mediated antitumor immune response. Sci Transl Med. 2021;13:eabj0835.
Yamashiro H, Yoshizaki S, Tadaki T, Egawa K, Seo N. Stimulation of human butyrophilin 3 molecules results in negative regulation of cellular immunity. J Leukoc Biol. 2010;88:757–67.
Chen S, Li Z, Huang W, Wang Y, Fan S. Prognostic and therapeutic significance of BTN3A proteins in tumors. J Cancer. 2021;12:4505–12.
Cano CE, Pasero C, De Gassart A, Kerneur C, Gabriac M, Fullana M, et al. BTN2A1, an immune checkpoint targeting Vγ9Vδ2 T cell cytotoxicity against malignant cells. Cell Rep. 2021;36:109359.
Girard P, Sosa Cuevas E, Ponsard B, Mouret S, Gil H, Col E, et al. Dysfunctional BTN3A together with deregulated immune checkpoints and type I/II IFN dictate defective interplay between pDCs and γδ T cells in melanoma patients, which impacts clinical outcomes. Clin Transl Immunol. 2021;10:e1329.
Gu S, Sachleben JR, Boughter CT, Nawrocka WI, Borowska MT, Tarrasch JT, et al. Phosphoantigen-induced conformational change of butyrophilin 3A1 (BTN3A1) and its implication on Vγ9Vδ2 T cell activation. P Natl Acad Sci USA. 2017;114:E7311–20.
Karunakaran MM, Willcox CR, Salim M, Paletta D, Fichtner AS, Noll A, et al. Butyrophilin-2A1 directly binds germline-encoded regions of the Vγ9Vδ2 TCR and Is essential for Phosphoantigen sensing. Immunity. 2020;52:487–498.e6.
Rigau M, Ostrouska S, Fulford TS, Johnson DN, Woods K, Ruan Z, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science. 2020;367:eaay5516 Available from: https://www.science.org/doi/10.1126/science.aay5516. Cited 2022 Sep 4.
Hsiao C-HC, Nguyen K, ** Y, Vinogradova O, Wiemer AJ. Ligand-induced interactions between butyrophilin 2A1 and 3A1 internal domains in the HMBPP receptor complex. Cell. Chem Biol. 2022;29:985–995.e5.
Cano CE, Pasero C, De Gassart A, Hoet R, Scotet E, Mortier E, et al. BTN2A, a New Immune-Checkpoint Targeting Vg9Vd2 T Cell Cytotoxicity. Blood. 2019;134:1044. Available from: Cited 2022 May 2. https://doi.org/10.1182/blood-2019-128658.
Vyborova A, Beringer DX, Fasci D, Karaiskaki F, van Diest E, Kramer L, et al. γ9δ2T cell diversity and the receptor interface with tumor cells. J Clin Invest. 2020;130:4637–51.
Payne KK, Mine JA, Biswas S, Chaurio RA, Perales-Puchalt A, Carmen M, et al. BTN3A1 governs antitumor responses by coordinating αβ and γδ T cells. Science. 2020;369:942–9 American Association for the Advancement of Science. Available from: https://www.science.org/doi/10.1126/science.aay2767. Cited 2022 Jul 14.
Huimin L, Shi T, Wang M, Li X, Yanzheng G, Zhang X, et al. B7-H3 inhibits the IFN-γ-dependent cytotoxicity of Vγ9Vδ2 T cells against colon cancer cells. Oncoimmunology. 2020;9:1748991.
Barjon C, Michaud H-A, Fages A, Dejou C, Zampieri A, They L, et al. IL-21 promotes the development of a CD73-positive Vγ9Vδ2 T cell regulatory population. Oncoimmunology. 2017;7:e1379642.
Guo Q, Zhao P, Zhang Z, Zhang J, Zhang Z, Hua Y, et al. TIM-3 blockade combined with bispecific antibody MT110 enhances the anti-tumor effect of γδ T cells. Cancer Immunol Immunother. 2020;69:2571–87.
Yang R, Shen S, Gong C, Wang X, Luo F, Luo F, et al. Bispecific antibody PD-L1 x CD3 boosts the anti-Tumor potency of the expanded Vγ2Vδ2 T cells. Front Immunol. 2021;12:654080.
Iwasaki M, Tanaka Y, Kobayashi H, Murata-Hirai K, Miyabe H, Sugie T, et al. Expression and function of PD-1 in human γδ T cells that recognize phosphoantigens. Eur J Immunol. 2011;41:345–55 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/eji.201040959. Cited 2022 Jul 17.
Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the efficiency of Vγ9Vδ2 T-Cell immunotherapy in Cancer. Front Immunol. 2018;9:800.
Hwang HJ, Lee JJ, Kang SH, Suh JK, Choi ES, Jang S, et al. The BTLA and PD-1 signaling pathways independently regulate the proliferation and cytotoxicity of human peripheral blood γδ T cells. Immun Inflamm Dis. 2021;9:274–87.
Tani-Ichi S, Wagatsuma K, Hara T, Cui G, Abe S, Miyachi H, et al. Innate-like CD27+CD45RBhigh γδ T cells require TCR signaling for homeostasis in peripheral lymphoid organs. J Immunol. 2020;204:2671–84.
Gertner-Dardenne J, Fauriat C, Orlanducci F, Thibult M-L, Pastor S, Fitzgibbon J, et al. The co-receptor BTLA negatively regulates human Vγ9Vδ2 T-cell proliferation: a potential way of immune escape for lymphoma cells. Blood. 2013;122:922–31. Available from: Cited 2022 Jul 17. https://doi.org/10.1182/blood-2012-11-464685.
Yang Z-Z, Kim HJ, Villasboas JC, Chen Y-P, Price-Troska T, Jalali S, et al. Expression of LAG-3 defines exhaustion of intratumoral PD-1 + T cells and correlates with poor outcome in follicular lymphoma. Oncotarget. 2017;8:61425–39 Impact Journals. Available from: https://www.oncotarget.com/article/18251/text/. Cited 2022 Jul 17.
Tomogane M, Sano Y, Shimizu D, Shimizu T, Miyashita M, Toda Y, et al. Human Vγ9Vδ2 T cells exert anti-tumor activity independently of PD-L1 expression in tumor cells. Biochem Biophys Res Commun. 2021;573:132–9 Available from: https://www.sciencedirect.com/science/article/pii/S0006291X21011451. Cited 2022 May 10.
Yang Q, Liu X, Liu Q, Guan Z, Luo J, Cao G, et al. Roles of mTORC1 and mTORC2 in controlling γδ T1 and γδ T17 differentiation and function. Cell Death Differ. 2020;27:2248–62.
Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. 2021;40:184 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8178863/. Cited 2022 Oct 4.
Schofield L, Ioannidis LJ, Karl S, Robinson LJ, Tan QY, Poole DP, et al. Synergistic effect of IL-12 and IL-18 induces TIM3 regulation of γδ T cell function and decreases the risk of clinical malaria in children living in Papua New Guinea. BMC Med. 2017;15:114.
Tirier SM, Mallm J-P, Steiger S, Poos AM, Awwad MHS, Giesen N, et al. Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nat Commun. 2021;12:6960.
Li X, Huimin L, Yanzheng G, Zhang X, Zhang G, Shi T, et al. Tim-3 suppresses the killing effect of Vγ9Vδ2 T cells on colon cancer cells by reducing perforin and granzyme B expression. Exp Cell Res. 2020;386:111719 Available from: https://www.sciencedirect.com/science/article/pii/S0014482719305981. Cited 2022 May 10.
van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, et al. NKG2A blockade potentiates CD8 T Cell Immunity induced by Cancer vaccines. Cell. 2018;175:1744–1755.e15 Elsevier. Available from: https://www.cell.com/cell/abstract/S0092-8674(18)31381-3. Cited 2022 Aug 24.
André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb Is a Checkpoint inhibitor that promotes anti-tumor Immunity by unleashing both T and NK cells. Cell. 2018;175:1731–1743.e13.
Peters C, Oberg H-H, Kabelitz D, Wesch D. Phenotype and regulation of immunosuppressive Vδ2-expressing γδ T cells. Cell Mol Life Sci. 2014;71:1943–60 Available from: https://springer.longhoe.net/article/10.1007/s00018-013-1467-1. Cited 2022 Jun 3.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
van Hall T, André P, Horowitz A, Ruan DF, Borst L, Zerbib R, et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer. 2019;7:263.
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29.
** Z, Ye W, Lan T, Zhao Y, Liu X, Chen J, et al. Characteristic of TIGIT and DNAM-1 expression on Foxp3+ γδ T cells in AML patients. BioMed Res Int. 2020, 2020:e4612952 Hindawi. Available from: https://www.hindawi.com/journals/bmri/2020/4612952/. Cited 2022 May 10.
Preillon J, Cuende J, Rabolli V, Garnero L, Mercier M, Wald N, et al. Restoration of T-cell effector function, depletion of Tregs, and direct killing of Tumor cells: The multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol Cancer Ther. 2021;20:121–31.
Lin A, Zhang H, Hu X, Chen X, Wu G, Luo P, et al. Age, sex, and specific gene mutations affect the effects of immune checkpoint inhibitors in colorectal cancer. Pharmacol Res. 2020;159:105028 Available from: https://www.sciencedirect.com/science/article/pii/S1043661820313360. Cited 2022 Oct 4.
Zhou C, Lin A, Cao M, Ding W, Mou W, Guo N, et al. Activation of the DDR pathway leads to the Down-regulation of the TGFβ pathway and a better response to ICIs in patients with metastatic urothelial carcinoma. Front Immunol. 2021;12:634741.
Tae Kon Kim, Esten N. Vandsemb, Roy S. Herbst, Lie** Chen. Adaptive immune resistance at the tumour site: mechanisms and therapeutic opportunities. Nat Rev Drug Discov. 2022;21:529–540. Nature Publishing Group. Available from: https://www.nature.com/articles/s41573-022-00493-5. Cited 2022 Aug 17
Jaiswal AR, Liu AJ, Pudakalakatti S, Dutta P, Jayaprakash P, Bartkowiak T, et al. Melanoma evolves complete immunotherapy resistance through the acquisition of a hypermetabolic phenotype. Cancer Immunol Res. 2020;8:1365–80. Available from: Cited 2022 Sep 18. https://doi.org/10.1158/2326-6066.CIR-19-0005.
Turiello R, Capone M, Morretta E, Monti MC, Madonna G, Azzaro R, et al. Exosomal CD73 from serum of patients with melanoma suppresses lymphocyte functions and is associated with therapy resistance to anti-PD-1 agents. J Immunother Cancer. 2022;10:e004043.
Lu J-C, Zhang P-F, Huang X-Y, Guo X-J, Gao C, Zeng H-Y, et al. Amplification of spatially isolated adenosine pathway by tumor-macrophage interaction induces anti-PD1 resistance in hepatocellular carcinoma. J Hematol Oncol. 2021;14:200.
Li C, Phoon YP, Karlinsey K, Tian YF, Thapaliya S, Thongkum A, et al. A high OXPHOS CD8 T cell subset is predictive of immunotherapy resistance in melanoma patients. J Exp Med. 2022;219:e20202084.
Lu H, Ma Y, Wang M, Shen J, Wu H, Li J, et al. B7-H3 confers resistance to Vγ9Vδ2 T cell-mediated cytotoxicity in human colon cancer cells via the STAT3/ULBP2 axis. Cancer Immunol Immunother. 2021;70:1213–26. Available from: Cited 2022 Sep 18. https://doi.org/10.1007/s00262-020-02771-w.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Liao D, Wang M, Liao Y, Li J, Niu T. A review of efficacy and safety of Checkpoint inhibitor for the treatment of acute myeloid leukemia. Front Pharmacol. 2019;10 Available from: https://www.frontiersin.org/article/10.3389/fphar.2019.00609. Cited 2022 May 10.
Li J, Smalley I, Chen Z, Wu J-Y, Phadke MS, Jamie K, et al. Single-cell Characterization of the Cellular Landscape of Acral Melanoma Identifies Novel Targets for Immunotherapy. Clin Cancer Res. 2022;28:2131–46. Available from: Cited 2022 Jun 4. https://doi.org/10.1158/1078-0432.CCR-21-3145.
Imai Y, Ayithan N, Xuesong W, Yuan Y, Wang L, Hwang ST. Cutting edge: PD-1 regulates Imiquimod-induced Psoriasiform dermatitis through inhibition of IL-17A expression by innate γδ-low T cells. J Immunol. 2015;195:421–5.
Kadekar D, Agerholm R, Viñals MT, Rizk J, Bekiaris V. The immune checkpoint receptor associated phosphatases SHP-1 and SHP-2 are not required for γδT17 cell development, activation, or skin inflammation. Eur J Immunol. 2020;50:873–9.
Li N, Wenwen X, Yuan Y, Ayithan N, Imai Y, Xuesong W, et al. Immune-checkpoint protein VISTA critically regulates the IL-23/IL-17 inflammatory axis. Sci Rep. 2017;7:1485.
Edwards SC, Hedley A, Hoevenaar WHM, Wiesheu R, Glauner T, Kilbey A, et al. PD-1 and TIM-3 differentially regulate subsets of mouse IL-17A-producing γδ T cells. J Exp Med. 2023;220:e20211431.
**ong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun. 2020;11:5084.
Gao J, Shi LZ, Zhao H, Chen J, **ong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404.e9 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867416311679. Cited 2022 Aug 19.
Choi J, Medikonda R, Saleh L, Kim T, Pant A, Srivastava S, et al. Combination checkpoint therapy with anti-PD-1 and anti-BTLA results in a synergistic therapeutic effect against murine glioblastoma. Oncoimmunology. 2021;10:1956142.
Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with Immune Checkpoint blockade. N Engl J Med. 2018;378:158–68.
Sullivan RJ, Weber JS. Immune-related toxicities of checkpoint inhibitors: mechanisms and mitigation strategies. Nat Rev Drug Discov. 2022;21:495–508.
Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38.
Tarhini AA, Zahoor H, Lin Y, Malhotra U, Sander C, Butterfield LH, et al. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer. 2015;3:39.
Alsaadi D, Shah NJ, Charabaty A, Atkins MB. A case of checkpoint inhibitor-induced celiac disease. J Immunother Cancer. 2019;7:203.
Eggesbø LM, Risnes LF, Neumann RS, Lundin KEA, Christophersen A, Sollid LM. Single-cell TCR sequencing of gut intraepithelial γδ T cells reveals a vast and diverse repertoire in celiac disease. Mucosal Immunol. 2020;13:313–21.
Badran YR, Shih A, Leet D, Mooradian MJ, Coromilas A, Chen J, et al. Immune checkpoint inhibitor-associated celiac disease. J Immunother Cancer. 2020;8:e000958.
Cheng M, Qian L, Shen G, Bian G, Xu T, Xu W, et al. Microbiota modulate tumoral immune surveillance in lung through a γδT17 immune cell-dependent mechanism. Cancer Res. 2014;74:4030–41.
Han J, Zhang S, Yi X, Pang Y, Xue Zhang YH, et al. Beneficial effect of antibiotics and microbial metabolites on expanded Vδ2Vγ9 T cells in hepatocellular carcinoma immunotherapy. Front Immunol. 2020;11 Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01380. Cited 2022 Jul 16.
Barisa M, Kramer AM, Majani Y, Moulding D, Saraiva L, Bajaj-Elliott M, et al. E. coli promotes human Vγ9Vδ2 T cell transition from cytokine-producing bactericidal effectors to professional phagocytic killers in a TCR-dependent manner. Sci Rep. 2017;7:2805.
Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22:516–23.
Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun. 2017;7:13839.
Xu L, Zou C, Zhang S, Chu TSM, Zhang Y, Chen W, et al. Resha** the systemic tumor immune environment (STIE) and tumor immune microenvironment (TIME) to enhance immunotherapy efficacy in solid tumors. J Hematol Oncol. 2022;15:87 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9264569/. Cited 2022 Oct 5.
Herrmann T, Fichtner AS, Karunakaran MM. An update on the molecular basis of Phosphoantigen recognition by Vγ9Vδ2 T cells. Cells. 2020;9:E1433.
Mu X, **ang Z, Xu Y, He J, Lu J, Chen Y, et al. Glucose metabolism controls human γδ T-cell-mediated tumor immunosurveillance in diabetes. Cell Mol Immunol. 2022;19:944–56.
Lopes N, McIntyre C, Martin S, Raverdeau M, Sumaria N, Kohlgruber AC, et al. Distinct metabolic programs established in the thymus control effector functions of γδ T cell subsets in tumor microenvironments. Nat Immunol. 2021;22:179–92.
Kim M, Min YK, Jang J, Park H, Lee S, Lee CH. Single-cell RNA sequencing reveals distinct cellular factors for response to immunotherapy targeting CD73 and PD-1 in colorectal cancer. J Immunother Cancer. 2021;9:e002503.
Taromi S, Firat E, Simonis A, Braun LM, Apostolova P, Elze M, et al. Enhanced AC133-specific CAR T cell therapy induces durable remissions in mice with metastatic small cell lung cancer. Cancer Lett. 2022;538:215697.
Laumont CM, Wouters MCA, Smazynski J, Gierc NS, Chavez EA, Chong LC, et al. Single-cell profiles and prognostic impact of Tumor-infiltrating lymphocytes Coexpressing CD39, CD103, and PD-1 in ovarian Cancer. Clin Cancer Res. 2021;27:4089–100.
Zhang T, Liu H, Jiao L, Zhang Z, He J, Li L, et al. Genetic characteristics involving the PD-1/PD-L1/L2 and CD73/A2aR axes and the immunosuppressive microenvironment in DLBCL. J Immunother Cancer. 2022;10:e004114.
Leem G, Park J, Jeon M, Kim E-S, Kim SW, Lee YJ, et al. 4-1BB co-stimulation further enhances anti-PD-1-mediated reinvigoration of exhausted CD39+ CD8 T cells from primary and metastatic sites of epithelial ovarian cancers. J Immunother Cancer. 2020;8:e001650.
Wang Y, Zhang H, Liu C, Wang Z, Wu W, Zhang N, et al. Immune checkpoint modulators in cancer immunotherapy: recent advances and emerging concepts. J Hematol Oncol. 2022;15:111.
Agrati C, Cimini E, Sacchi A, Bordoni V, Gioia C, Casetti R, et al. Activated V gamma 9V delta 2 T cells trigger granulocyte functions via MCP-2 release. J Immunol. 2009;182:522–9.
Devilder M-C, Maillet S, Bouyge-Moreau I, Donnadieu E, Bonneville M, Scotet E. Potentiation of antigen-stimulated V gamma 9V delta 2 T cell cytokine production by immature dendritic cells (DC) and reciprocal effect on DC maturation. J Immunol. 2006;176:1386–93.
Whiteside SK, Snook JP, Williams MA, Weis JJ. Bystander T cells: a balancing act of friends and foes. Trends Immunol. 2018;39:1021–35 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6269193/. Cited 2022 Dec 7.
Lee H, Jeong S, Shin E-C. Significance of bystander T cell activation in microbial infection. Nat Immunol. 2022;23:13–22.
Paul S, Lal G. Regulatory and effector functions of gamma–delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int J Cancer. 2016;139:976–85 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ijc.30109. cited 2022 Jul 17.
Meier SL, Satpathy AT, Wells DK. Bystander T cells in cancer immunology and therapy. Nat Cancer. 2022;3:143–55.
Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-Delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol. 2020;11:1347.
Ye X, Deng X, Wen J, Li Y, Zhang M, Cai Z, et al. Folate receptor-alpha targeted 7x19 CAR-γδT suppressed triple-negative breast cancer xenograft model in mice. J Oncol. 2022;2022:2112898.
Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human Gamma Delta T cells: enhanced Cytotoxicity with retention of cross presentation. Mol Ther. 2018;26:354–65.
Rotolo R, Leuci V, Donini C, Cykowska A, Gammaitoni L, Medico G, et al. CAR-based strategies beyond T lymphocytes: integrative opportunities for Cancer adoptive immunotherapy. Int J Mol Sci. 2019;20:2839.
Fisher J, Sharma R, Don DW, Barisa M, Hurtado MO, Abramowski P, et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci Signal. 2019;12:eaax1872.
Harrer DC, Simon B, Fujii S-I, Shimizu K, Uslu U, Schuler G, et al. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer. 2017;17:551.
Harrer DC, Dörrie J, Schaft N. Chimeric antigen receptors in different Cell types: New vehicles join the race. Hum Gene Ther. 2018;29:547–58.
** C, Ma J, Ramachandran M, Yu D, Essand M. CAR T cells expressing a bacterial virulence factor trigger potent bystander antitumour responses in solid cancers. Nat Biomed Eng. 2022;6:830–41.
Ferry GM, Agbuduwe C, Forrester M, Dunlop S, Chester K, Fisher J, et al. A simple and robust single-step method for CAR-Vδ1 γδT Cell expansion and transduction for Cancer immunotherapy. Front Immunol. 2022;13:863155.
Ganapathy T, Radhakrishnan R, Sakshi S, Martin S. CAR γδ T cells for cancer immunotherapy. Is the field more yellow than green? Cancer Immunol Immunother. 2022; Available from: Cited 2022 Dec 13. https://doi.org/10.1007/s00262-022-03260-y.
Thedrez A, Harly C, Morice A, Salot S, Bonneville M, Scotet E. IL-21-mediated potentiation of antitumor cytolytic and proinflammatory responses of human V gamma 9V delta 2 T cells for adoptive immunotherapy. J Immunol. 2009;182:3423–31.
Zumwalde NA, Sharma A, Xuequn X, Ma S, Schneider CL, Romero-Masters JC, et al. Adoptively transferred Vγ9Vδ2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight. 2017;2 American Society for Clinical Investigation. Available from: https://insight.jci.org/articles/view/93179. Cited 2022 Jul 17.
Nada MH, Wang H, Hussein AJ, Tanaka Y, Morita CT. PD-1 checkpoint blockade enhances adoptive immunotherapy by human Vγ2Vδ2 T cells against human prostate cancer. OncoImmunology. 2021;10:1989789. Taylor & Francis. Available from: Cited 2022 Jun 3. https://doi.org/10.1080/2162402X.2021.1989789.
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (2018A030313846 and 2021A1515012593), the Science and Technology Planning Project of Guangdong Province (2019A030317020) and the National Natural Science Foundation of China (81802257, 81871859, 81772457, 82172750 and 82172811). GuangDong Basic and Applied Basic Research Foundation (Guangdong - Guangzhou Joint Fouds) (2022A1515111212). This work was supported by Key Research and Development Projects of Sichuan Science and Technology (2022YFS0221, 2022YFS0074, 2022YFS0156 and 2022YFS0378). Sichuan Provincial People's Hospital Hospital Young Talent Fund (2021QN08).
Author information
Authors and Affiliations
Contributions
PL, JZ and XC conceived the study content and provided constructive guidance. ZFG, AQL, YFB and AMJ collected and organized the related references, and visualized the figures. ZFG wrote the manuscript. AQL corrected the manuscript. ZFG, AQL, YFB and AMJ made significant revisions to the manuscript. AQL and CZZ completed the additional analyses and prepared related figures. QC and ZQL complemented the manuscript. ZFG and AQL did the English editing, revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Heatmap of γδT cells in TCGA dataset. The heatmap is showing characteristic distributions of pan-γδT cells and other γδT-cell subgroups (column labels) in different types of cancers (row labels).
Additional file 2: Fig. S2.
Heatmap of γδT cells in GTEx dataset. The heatmap is showing characteristic distributions of pan-γδT cells and other γδT-cell subgroups (column labels) in different types of tissues (row labels).
Additional file 3: Fig. S3.
Anatomical heatmaps of γδT cells in TCGA dataset. Anatomical heatmaps exhibit enrichment scores of γδT cells and other subtypes, including pan-γδT cell, Vγ9Vδ2T cells, cytotoxic γδT cells, IFN-γ-producing γδT cells, γδNKT cells, type2-like γδT cells, γδT17 cells and γδTregs across given anatomic locations.
Additional file 4: Fig. S4.
Anatomical heatmaps of γδT cells in GTEx dataset. Anatomical heatmaps exhibit enrichment scores of γδT cells and other subtypes, including pan-γδT cell, Vγ9Vδ2T cells, cytotoxic γδT cells, IFN-γ-producing γδT cells, γδNKT cells, type2-like γδT cells, γδT17 cells and γδTregs across given anatomic locations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, Z., Bai, Y., Lin, A. et al. Gamma delta T-cell-based immune checkpoint therapy: attractive candidate for antitumor treatment. Mol Cancer 22, 31 (2023). https://doi.org/10.1186/s12943-023-01722-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01722-0