
Abstract
Background
As a highly heterogeneous tumor, non-small cell lung cancer (NSCLC) is famous for its high incidence and mortality worldwide. Smoking can cause genetic changes, which leading to the occurrence and progress of NSCLC. Nevertheless, the function of smoking-related genes in NSCLC needs more research.
Methods
We downloaded transcriptome data and clinicopathological parameters from Gene Expression Omnibus (GEO) databases, and screened smoking-related genes. Lasso regression were applied to establish the 7-gene signature. The associations between the 7-gene signature and immune microenvironment analysis, survival analysis, drug sensitivity analysis and enriched molecular pathways were studied. Ultimately, cell function experiments were conducted to research the function of FCGBP in NSCLC.
Results
Through 7-gene signature, NSCLC samples were classified into high-risk group (HRG) and low-risk group (LRG). Significant difference in overall survival (OS) between HRG and LRG was found. Nomograms and ROC curves indicated that the 7-gene signature has a stable ability in predicting prognosis. Through the analysis of immune microenvironment, we found that LRG patients had better tumor immune activation. FCGBP showed the highest mutation frequency among the seven prognostic smoking related genes (LRRC31, HPGD, FCGBP, SPINK5, CYP24A1, S100P and FGG), and was notable down-regulated in NSCLC smokers compared with non-smoking NSCLC patients. The cell experiments confirmed that FCGBP knockdown promoting proliferation, migration, and invasion in NSCLC cells.
Conclusion
This smoking-related prognostic signature represents a promising tool for assessing prognosis and tumor microenvironment in smokers with NSCLC. The role of FCGBP in NSCLC was found by cell experiments, which can be served as diagnostic biomarker and immunotherapy target for NSCLC.
Similar content being viewed by others

Introduction
Ranking second in cancer incidence and first in cancer-related mortality, lung cancer causes about 700,000 deaths in China per year [1, 2]. Among lung cancer pathological subtypes, non-small cell lung cancer (NSCLC) was the most common pathological subtype, which accounts for almost 85% [3]. With the widespread application of new immunotherapy, surgical treatment and chemotherapy, NSCLC patients’ 5-year survival rate has gotten notable upgraded [4]. However, NSCLC patients are often diagnosed in the advanced stage, which is major cause of treatment failure and poor prognosis [5]. Therefore, exploring novel prognostic assessment and treatment options of NSCLC is indispensable.
Smoking is considered to be the most preventable cause of tumor occurrence and death [6]. The number of cancer deaths caused by smoking accounts for approximately one-third of all cancer deaths every year [7]. Numerous studies have proven that smoking will induce many cancers’ occurrence [8,9,10,11,12,13]. The change in smoking is paralleled by a change in the incidence of NSCLC [14]. Tobacco smoke contains more than 60 known or suspected carcinogens. Long-term smoking will inhibit the DNA repair mechanism and cause genetic changes, which lead to the occurrence and progression of cancer [15]. Due to gene changes precede obvious histopathological changes in tumor detection, it is urgent to identify the genetic changes of smoking and construct a new biomarker that can be used to stratify patients with NSCLC at the early stage. In recent years, it is a trend to construct new tumor biomarkers for NSCLC, and remarkable achievements have been achieved [16,17,18,19].
Herein, we identified differentially expressed smoking-related genes and constructed smoking-related prognostic signature to predict prognosis, TME, drug sensitivity and immunotherapeutic effect of NSCLC patients with smoking history. In addition, among differentially expressed smoking-related genes, we demonstrated the function of the highest mutation frequency gene FCGBP in NSCLC by cell experiments. Our results may yield a robust biomarker for assessing prognosis and tumor microenvironment of NSCLC patients with smoking history and immunotherapy response.
Materials and methods
Date preparation
In this study, NSCLC gene expression datasets were downloaded three cohorts (GSE50081, GSE68465 and GSE72094) from Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). Eliminating the data with incomplete smoking history and survival information, we obtained 106 NSCLC patients without smoking history and 736 NSCLC samples with smoking history. In addition, the copy number variation (CNV) frequency of somatic mutations was downloaded from the Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov/) for genetic mutation analysis.
Identification of differentially expressed smoking-related genes
To identifying smoking-related genes, we applied the ‘limma’ R package to identify DEGs between NSCLC smokers and NSCLC patients without smoking history. Genes met the conditions of log2 | FC |>1 and FDR < 0.05 are considered significant.
Construction and validation of the prognostic signature
Applying univariate Cox regression analysis, prognostic smoking-related genes were filtered (P < 0.05). Then, using the R package ‘glmnet’, the least absolute contraction and selection operator (LASSO) Cox regression algorithm to minimize the risk of over fitting combining selected factors. Then, a 7-gene signature was constructed based on the screened prognostic smoking-related genes. Risk score was computed by the formula: risk score=∑ (gene × coefficient). NSCLC smokers were separated into high-risk group and low-risk group by the mid-value of the risk scores. To verify the 7-gene signature, external validation group by merging GSE29016 and GSE102287 was applied to test the performance of the signature in forecasting clinical results.
Gene Set Enrichment Analysis (GSEA) and single sample Gene Set Enrichment analysis (ssGSEA)
Gene set “c2.cp.kegg.v7.4.symbols.gmt” from the MSigDB database (https://www.gsea-msigdb.org) was obtained to conduct GSEA. ssGSEA is an extension of the GSEA method, was applied to calculate the infiltration degree of 23 kinds of immune cells in each sample. Using the ‘GSEABase’ and ‘GSVA’R package, Differential expression related pathways and immune cells between LRG and HRG were identified. We simulated calculations 1000 times to obtain stable final data.
Immune function analysis and immune checkpoint analysis
‘limma’, ‘GSVA’, ‘GSEABase’, ‘pheatmap’ and ‘reshape2’ R package were used to perform immune function analysis. The expression of immune checkpoints was determined through R package ‘limma’, ‘ggplot2’, ‘ggpubr’ and ‘reshape2’.
Drug sensitivity analysis
The R package ‘gpubr’ and ‘pRRophetic’ R packages were used by us to explore chemotherapeutic drugs in different risk groups of NSCLC patients by calculating the half-inhibitory concentration (IC50) values of different drugs.
NSCLC cell culture
Human NSCLC cell lines (A549 and NCI-H23) were purchased from the Shanghai Cell Bank, Chinese Academy of Sciences. NSCLC cell lines were cultured in the medium which consists of RPMI-1640 (Gibco, USA), 10% fetal bovine serum (Gibco, USA), 0.1 mg/ml streptomycin (Gibco, USA) and 100U/ml penicillin (Gibco, USA) and maintained under 37 °C at 5% CO2 atmosphere. Lentivirus targeting FCGBP knockdown was constructed by Hanbio Co. LTD (Shanghai, China). Target Seq: GGTGATCCATTCTGACTATGC.
qPCR
The RNA-easy Isolation Reagent (R701, Vazyme, China) was used to extract total RNA from A549 and NCI-H23 cells. The HiScript III 1st Strand cDNA Synthesis Kit (R312, Vazyme, China) was used to synthetise cDNA. qPCR was applied to verity the knockdown efficiency of the synthesized shRNAs. qPCR was conducted by ChamQ Universal SYBR qPCR Master Mix (Q711, Vazyme, China) on the QuantStudio 5 Real-Time PCR Systems (A28569, Thermo Fisher Scientific, USA). Relative quantification was gotten by the 2−ΔΔCT method. The primer used was as follows:
FCGBP-Forward primer: 5ʹ-GCAGTGAGTTCTCGTATGCTGAA-3ʹ;
FCGBP-Reverse primer: 5ʹ-GAAGGTGAGCAGTCCCAAGTT-3ʹ.
Cell counting Kit-8 (CCK-8) assay
CCK8 assay was applied to detect A549 and NCI-H23 cells proliferation ability. 2000 transfected cells were inoculated in 96-well plates and added 10 µL of CCK-8 reagent (BS350A, Biosharp, China). The absorbance of cells at 450 nm was detected every 24 h for 5 days.
Transwell assay
Precoat the Cell Culture Insert (353,097, BD Falcon, USA) with or without Matrix gel (356,234, Corning, USA) to evaluate the invasion or migration ability of cells respectively. 50,000 transfected cells were inoculated into the upper well. Adding serum free alkaline gel to the upper chamber, then we added the medium containing 20% FBS to the lower chamber in both assays. After 24 h of culture, the cells on the insert were fixed with polyformaldehyde, and finally dyed with crystal violet.
Clone formation assay
500 transfected A549 and NCI-H23 cells were inoculated on the 6-well plate respectively. After incubating for 9 days, cells were fixed with paraformaldehyde and stained with crystal violet.
Cell cycle and apoptosis analysis
Steps of cell cycle experiment: First, we fixed the cells in 70% alcohol overnight at 4 °C, washed them, and dyed them with propidium iodide (PI) containing RNase A (CA1510, Solarbio, China). Finally, the percentage of nsclc cell cycle phases (G0/G1, S and G2/M phases) was detected by flow cytometry (FACSCalibur, BD, USA). For apoptosis assay, cells washed with pre-cooled PBS and stained with Annexin V-Alexa Fluor 647/PI Apoptosis Assay Kit (FMSAV647, Fcmacs, China) by the manual. We used flow cytometry (FACSCalibur, BD, USA) to analyze the cells for apoptosis.
Statistical analysis
The data is dealt with GraphPad Prism software and expressed as mean ± SD. The R 4.2.0 was applied to perform data processing.
Results
Identification of differentially expressed smoking-related genes in NSCLC
The flowchart of the research is shown in Fig. 1. 842 NSCLC cases that came from GSE50081, GSE68465 and GSE72094. Deleting unqualified cases, we obtained 106 NSCLC patients without smoking history and 736 NSCLC patients with smoking history. The mRNA expression profiles of genes between NSCLC samples with smoking history and NSCLC samples without smoking history was analyzed. With the condition of p < 0.05 and |log2 FC| ≥ 1, there are 20 smoking-related genes were differentially expressed. Among these smoking-related genes, 11 genes were upregulated and 9 genes were downregulated (Fig. 2A). The expression landscape of the 20 differentially expressed smoking-related genes in smoking group and non-smoking group were illustrated in a heatmap (Fig. 2B).

Research flow chart

Identifying of differentially expressed smoking-related genes from GSE50081, GSE68465 and GSE72094. (A) Volcano plot of differentially expressed smoking-related genes by the conditions of log2 | FC |>1 and FDR < 0.05. (B) The heatmap of differentially expressed smoking-related genes expression landscape in smoking and non-smoking groups
Construction of prognostic signature based on prognostic smoking-related genes for patient with smoking history
Applying univariate Cox regression analysis, 7 genes (LRRC31, HPGD, FCGBP, SPINK5, CYP24A1, S100P and FGG) out of the 20 differentially expressed smoking-related genes were significantly correlated with overall survival (OS) of NSCLC patients with smoking history in GSE50081, GSE68465 and GSE72094 cohort (Fig. 3A). Lasso Cox regression was used to identify the key genes with the best prognostic value by reducing the dimension, and the relative coefficient of smoking related genes was calculated (Fig. 3B-C). The risk score is obtained by the following formula. Risk score = LRRC31 exp. * (-0.071490275115065) + HPGD exp. * (-0.0338294194692034) + FCGBP exp. * (-0.0289843021911047) + SPINK5 exp.* (-0.0289133244265656) + CYP24A1 exp. * 0.0572743744393659 + S100P exp. * 0.0237236238563723 + FGG exp. * 0.0335046687925319. Then, by the median value of risk score, NSCLC patients with smoking history were separated in low-risk group (LRG) and high-risk group (HRG) (Fig. 3D). The clinical data of HRG and LRG patients are presented in Table 1. Then, conducting survival analysis, we found that HRG patients had a worse OS than those in LRG (Fig. 3E). A heatmap visualized the distribution of LRRC31, HPGD, FCGBP, SPINK5, CYP24A1, S100P and FGG in two groups (Fig. 3F). Next, univariate and multivariate Cox regression analyses demonstrated that the 7-gene risk signature is an independent prognostic indicator for predicting prognosis of NSCLC patients with smoking history (Fig. 3G-H).

Establishing the 7-gene signature. (A) Forest plot of seven prognostic smoking related genes screened using Cox univariate regression analysis. (B, C) Cvift and lambda curves of LASSO regression applied with minimum criteria. (D) Risk scores and survival status of patients in two risk groups. (E) K-M curves of the OS of HRG and LRG. (F) Heatmap of the expression of seven prognostic smoking-related genes in HRG and LRG. (G, H) Univariate and multivariate Cox regression analysis of the 7-gene risk signature
Clinical information evaluation of prognostic risk signature
ROC curve was first conducted to verify the reliability of the prognostic risk signature. Comparing different clinical parameters, the ROC value of the 7-gene risk signature was 0.644 (Fig. 4A). The area under curve (AUC) of predicting 1-, 2-, and 3-year survival rates were 0.644, 0.659, and 0.651, respectively (Fig. 4B). Meanwhile, we analyzed the clinical parameters of the risk group and constructed a heatmap based on the 7 prognostic smoking-related genes. (Fig. 4C). Applying Kaplan-Meier (K-M) survival analysis, we have stratified the clinical characteristics of HRG and LRG. Firstly, NSCLC patients with smoking history were divided into two stages by age: old stage (age > 65) and young stage (age ≤ 65). In two stages, HRG had worse OS compared with the LRG (Supplementary Fig. 1A, B). Next, according to gender, we found that HRG had a worse OS in both male and female groups (Supplementary Fig. 1C, D). Similarly, according to stage grade, samples of stage 1 and stage 2 were analyzed (Supplementary Fig. 1E, F). To sum up, stratified survival analysis indicated that the 7-gene risk signature had stable ability to predict prognosis in different clinical characteristics.

Clinical value analysis of the 7-gene risk signature. (A) ROC curves of clinical parameters and risk scores. (B) ROC curves of the 7-gene risk signature predicting the OS of 1-, 2-, and 3-year. (C) Heatmap illustrating correlations between clinical parameters and risk groups
Construction and evaluation of Nomogram
In order to assess the risk of NSCLC patients with smoking history more accurately, we constructed a nomogram combining clinical feature and prognostic risk signature. According to the sum of relevant factors, the prediction probability of 1-, 3-, and 5-year survival rates were 0.925, 0.752, and 0.62, respectively (Fig. 5A). The calibration chart also showed the good prediction accuracy of the nomogram (Fig. 5B). Using ROC analysis, the AUC result of the nomogram was 0.722, which showed the best prediction accuracy compared with others clinical parameters (Fig. 5C). Cox univariate regression analysis indicated that the nomogram had relevance with NSCLC smokers’ OS (Fig. 5D). Moreover, according to multivariate Cox regression analysis, we found that the nomogram was an independent prognostic factor for forecasting the OS rates of NSCLC patients with smoking history in GSE50081, GSE68465 and GSE72094 cohort (Fig. 5E).

Construction and evaluation of nomogram. (A) Nomogram predicting 1-, 3-, and 5-year OS rate of NSCLC smokers. (B) Calibration curve for assessing the accuracy of the nomogram in predicting 1-, 3-, and 5-year OS rate. (C) ROC curves of clinical characteristics and the nomogram. (D, E) Univariate and multivariate Cox regression analysis of the nomogram. “*” P < 0.05, “**” P < 0.01, and “***” P < 0.001
Immune Microenvironment of 7-gene risk signature
Applying GSEA, we found that patients in LRG have better enrichment of immune activation and enrichment of patients in HRG more inclined to cellular mechanism such as cell cycle and RNA degradation (Fig. 6A). Subsequently, applying ‘limma’, ‘GSVA’, ‘GSEABase’, ‘pheatmap’ and ‘reshape2’ R package immune function analysis was performed to research difference of enrichment of immune pathways between HRG and LRG. At the same time, we visualized the enrichment results through heatmaps. In the heatmap, we found that tumor suppressor immune pathways such as APC co inhibition, Type II IFN response, Type I IFN response and HLA were significantly activated in LRG (Fig. 6B). In order to investigate the immune microenvironment, the infiltration degree of 23 immune cells in HRG and LRG was detected. Activated CD4 T cell, CD56dim natural killer cell, neutrophil and type 2 T helper cell, which interrelate with immunosuppression, had higher infiltration degree in HRG. In the other side, eleven kinds of immune cells interrelate with tumor immune activation were widely distributed in LRG, which indicate a better immune response of patients in LRG (Fig. 6C). Overall, LRG patients have better tumor immune activation, which explains why patients in LRG have better OS than those in HRG. In order to obtain better immunotherapy effect for patients in HRG, we explored the difference of immune checkpoints in two risk groups. 23 common immune checkpoint genes were found to be statistically different between the two risk groups. Patients in HRG had high expression of 11 kinds of immune checkpoint genes, including immunotherapy targets that have been proved effective in the treatment of lung cancer, such as PDCD1 (PD-1), TNFRSF8 (CD30), IDO1 and LAG3. This discovery provides new possibilities for HRG patients to provide more effective immunotherapy strategies (Fig. 6D).

Immune-related analysis of HRG and LRG. (A) GSEA analysis showed significant pathway enrichment between HRG and LRG (B) The heatmap of immune function analysis results between HRG and LRG. (C) Boxplot of the infiltration degree of immune cells infiltration. (D) Differences in expression levels of 23 immune checkpoints between HRG and LRG. “*” P < 0.05, “**” P < 0.01, and “***” P < 0.001
Drug sensitivity analysis
To further explore the connection between the 7-gene risk signature and clinical medical treatment, we used the pRRophetic algorithm to assess drug susceptibilities by the half-maximal inhibitory concentration (IC50). By comparing the IC50 levels of HRG and LRG, 18 kinds of drugs were obtained. The analysis results show that patients in HRG have a higher sensitivity to common chemotherapy and immunotherapy drugs including sorafenib, doxorubicin, imatinib, midostaurin, pyrimethamine and vinorelbine, which make contributions to better prognosis of LRG patients (Supplementary Fig. 2A-M). Furthermore, rapamycin, MG-132, erlotinib, WZ-1-84 and Z-LLNle-CHO may be become new drugs to ameliorate the OS of LRG patients (Supplementary Fig. 2N-R).
Validation of the 7-gene risk signature by the GSE29016 and GSE102287
To verify the stability of the 7-gene risk signature ‘s ability to predict the prognosis of NSCLC smokers, we established an external validation group by merging GSE29016 and GSE102287 and deleting unqualified patient data. The risk score of patients in external verification were gotten by the same formula. Same as mentioned above, HRG and LRG were distinguished according to the median of risk scores. Figure 7A visualized risk score and survival status’ distribution of patients in external verification. Survival analysis indicated that HRG patients had a worse OS compared the LRG group as expected (Fig. 7B). Univariate (Fig. 7C) and multivariate (Fig. 7D) cox regression analysis result showed that the 7-gene risk signature is an independent prognostic factor for predicting the OS of patients in external verification. ROC curve indicated that this risk signature predicted prognosis more accurately than other clinicopathological features in external verification (Fig. 7E). A time-dependent ROC curve showed the AUCs at 1, 2, and 3 years were 0.767, 0.660, and 0.695, respectively (Fig. 7F). The expression of seven prognostic smoking-related genes (LRRC31, HPGD, FCGBP, SPINK5, CYP24A1, S100P and FGG) in patients of external verification was shown by the heatmap (Fig. 7G). Ultimately, nomogram was constructed and the mortality rate of the patient in 1, 3 and 5 years was assessed to be 0.923, 0.777 and 0.641 (Fig. 7H). In summary, this 7-gene risk signature effectively predict the risk of NSCLC smokers in external verification, demonstrating the robust and stable predictive ability of this smoking-associated risk signature.

Validation of this 7-gene risk signature by the GSE29016 and GSE102287. (A) Risk score and survival status of NSCLC smokers in the GSE29016 and GSE102287. (B) Survival analysis of LRG and HRG. (C) Univariate Cox regression analysis and (D) multivariate Cox regression analysis of 7-gene risk signature. (E) ROC curves of clinical characteristics and 7-gene risk signature. (F) ROC curves indicate 7-gene risk signature forecasting the 1-, 2-, and 3-year OS. (G) Heatmap of the seven genes of LRG and HRG in external verification. (H) Nomogram based on 7-gene risk signature, age, gender, and TNM stage
Genetic Mutation and Survival Analysis of FCGBP
Owning to the close relationship between smoking and gene mutation, genetic mutation analysis was applied to explore mutation frequency of these seven prognostic smoking-related genes in NSCLC. With 12.85% mutation frequency in 1121 samples, 144 samples had prognostic smoking-related genes mutations. Among seven prognostic smoking-related genes, FCGBP carried the highest mutation frequency at 9%, so we further conducted further analysis on FCGBP (Supplementary Fig. 3A). Performing expression level analysis in patients, the expression level of FCGBP was found significantly downregulated in NSCLC patients with smoking history compared with non-smoking NSCLC patients (Supplementary Fig. 3B). Survival analysis indicated that the low expression of FCGBP interrelate with the deterioration of prognosis in smokers with NSCLC (Supplementary Fig. 3C).
FCGBP Knockdown Promoting Proliferation, Migration, and Invasion in NSCLC Cells
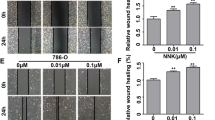
Owning to smoking can cause FCGBP mutation and reduce the expression, we transfected FCGBP knockdown plasmids into A549 and NCI-H23 cells to investigate the effect of FCGBP knockdown in NSCLC cells (Fig. 8A). CCK-8 assays indicated that knockdown of FCGBP significantly promoted the growth rate of A549 and NCI-H23 cells (Fig. 8B). Compared with the control group, the colony forming units were significantly increased after FCGBP knockdown in A549 and NCI-H23 cells (Fig. 8C, D). Next, flow cytometry cell cycle analysis showed S-phase cells increased after FCGBP knockdown in A549 and NCI-H23 cells (Fig. 8E, F). In addition, flow cytometry demonstrated that apoptosis rate of FCGBP knockdown NSCLC cells significantly decreased (Fig. 8G, H). Applying transwell assays, we found that knockdown of FCGBP significantly strengthened the migration and invasion abilities of A549 and NCI-H23 cells (Fig. 8I-L). These experimental results support the above bioinformatics analysis conclusions and provide solid evidence for the rationality of selecting this prognostic model (Fig. 9).

Exploring the effects of FCGBP knockout on LUAD cells through cell experiments (A) qPCR to investigate the knockdown efficiency of FCGBP in A549 and NCI-H23 cells. (B) CCK-8 assay to assess the effect of knockdown of FCGBP on the proliferation ability of A549 and NCI-H23 cells. (C, D) Compared with the siRNA negative control (NC) group, colony formation assay was applied to evaluate the effect of knockout of FCGBP of proliferative capacity in A549 and NCI-H23 cells. (E-H) Flow cytometry analysis was used to assess the effect of FCGBP knockdown on cell cycle and apoptosis. (I-L) Transwell analysis of migration and invasion ability of FCGBP knockdown in NSCLC cells

Summary diagram of this research work
Discussion
As the most common subtype of lung cancer with high heterogeneity, NSCLC was known for its high mortality rate overall the world [20]. Due to clinical symptoms of NSCLC appear late, many patients are diagnosed at an advanced stage, thus missing the best treatment opportunity [21, 22]. Owning to gene changes precede obvious histopathological changes in cancer detection, there is an urgent need for a biomarker based on genetic aspect to predict the early survival prognosis, tumor microenvironment, immunotherapy targets and drug sensitivity of NSCLC [23, 24].
At present, smoking is regarded as the main cause of lung cancer, and nearly 90% of lung cancer can be attributed to smoking [25]. Smoking will induce multiple DNA damage, inhibit DNA repair and cause gene changes, thus promoting the occurrence and development of lung cancer [26]. Many studies have shown that NSCLC of non-smokers and NSCLC of smokers have different molecular biology [27, 28]. The development of gene sequencing technology makes it possible to identify some potential genes with predictive value in NSCLC patients with smoking history and non-smoking NSCLC patients [29, 30]. However, there are few reliable biomarkers to help diagnosis. Therefore, it is necessary to identify smoking-related genes and construct robust risk signature to help early diagnosis of NSCLC patients.
In this research, we identified 20 differentially expressed smoking-related genes by analyzing the mRNA expression profiles of genes between NSCLC patients with smoking history and NSCLC samples without smoking history. In order to accurately quantify and assess smoking-related genes in NSCLC, Lasso Cox regression was applied to build a prognostic risk model. By calculating the risk score, samples were classified into HRG and LRG. Patients in HRG showed worse outcomes (P < 0.001). ROC curve and nomogram are regarded as reliable tools to forecasting the prognosis of cancer [31,32,33]. ROC curves and nomogram demonstrated the 7-gene signature had high accuracy in farecasting the prognosis of NSCLC patients with smoking history. Furthermore, stratified analysis showed that the 7-gene prognostic signature maintained a stable predictive ability in terms of age, gender and stage. In addition, higher immune cell infiltration that promote tumor immunity was found in LRG, such as CD56 bright natural killer cell, eosinophil, mast cell, and T follicular helper cell [34,35,36,37]. The analysis of immune pathway also indicated that patients in LRG have better antitumor immune activation, which contribute to better outcomes. A significant connection between the risk signature and immunotherapy was found, which indicated this signature provides new possibilities for HRG patients to provide more effective immunotherapy strategies. Last but not least, we conducted IC50 analysis to evaluate drug sensitivity and screen drugs to provide reference for future treatment.
The risk signature is composed of seven prognostic smoking-related genes (LRRC31, HPGD, FCGBP, SPINK5, CYP24A1, S100P and FGG). Among these genes, LRRC31, HPGD, FCGBP and SPINK5 are served as protected factors. In the other hand, CYP24A1, S100P and FGG are found as risk factors. Previous research confirmed the accuracy of our research. LRRC31 was found to be a DNA repair suppressor that can target cancer radiation to increase sensitivity [38]. Overexpression of HPGD inhibited the proliferation, migration and anchoring growth of cervical cancer cells [39]. SPINK5 plays a tumor inhibitor role in NSCLC by negatively regulating PSIP1 [40]. Low expression of CYP24A1 is correlated with poor prognosis in breast cancer [41]. S100P was found to increase the migration and invasion of cancer cells in lung cancer [42]. FGG regulates the expression of SLUG and ZEB1, and promotes the migration and invasion of hepatocellular carcinoma cells through EMT signal pathway [43]. IgG Fc-binding protein (FCGBP) was found to be closely related to mutations caused by smoking [44]. Applying genetic mutation analysis, FCGBP had the highest mutation frequency among seven prognostic smoking-related genes, which is identified as the key gene of smoking mutation. FCGBP has been proven to participate in intestinal tumor immunity [45]. However, the role of FCGBP in NSCLC has not been researched. We found that FCGBP was significantly downregulated in NSCLC patients with smoking history compared with non-smoking NSCLC patients. Cell experiments demonstrated that FCGBP knockdown promoting proliferation, migration, and invasion in A549 and NCI-H23 cell lines. This shows that the low expression of FCGBP caused by smoking can promote the progress of NSCLC, which provides a novel screening biomarker and treatment targets for NSCLC smokers.
This study also had some noteworthy limitations. On the one hand, more NSCLC samples are needed to maintain the reliability of smoking-related prognostic signature. On the other hand, further research on upstream and downstream pathway of FCGBP in NSCLC is necessary.
Conclusion
To sum up, we built a prognostic risk model based on seven prognostic smoking-related genes, which can accurately evaluate the prognosis, immunotherapy, drug sensitivity and tumor microenvironment of NSCLC patients. Importantly, the role of FCGBP in NSCLC was detected by cell experiments, which provides a screening biomarker and therapeutic target for NSCLC.
Data availability
Not applicable.
Code availability
Not applicable.
References
**a C, Dong X, Li H, Cao M, Sun D, He S, Yang F, Yan X, Zhang S, Li N, Chen W. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl). 2022;135:584–90.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54.
Horvath L, Thienpont B, Zhao L, Wolf D, Pircher A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC) - novel approaches and future outlook. Mol Cancer. 2020;19:141.
Nooreldeen R, Bach H. Current and Future Development in Lung Cancer diagnosis. Int J Mol Sci 2021, 22.
Fillon M. Pairing smoking cessation with lung cancer screening may save lives. CA Cancer J Clin. 2021;71:283–4.
Balogh EP, Dresler C, Fleury ME, Gritz ER, Kean TJ, Myers ML, Nass SJ, Nevidjon B, Toll BA, Warren GW, Herbst RS. Reducing tobacco-related cancer incidence and mortality: summary of an institute of medicine workshop. Oncologist. 2014;19:21–31.
Guenel P, Chastang JF, Luce D, Leclerc A, Brugere J. A study of the interaction of alcohol drinking and tobacco smoking among French cases of laryngeal cancer. J Epidemiol Community Health. 1988;42:350–4.
Gormley M, Dudding T, Sanderson E, Martin RM, Thomas S, Tyrrell J, Ness AR, Brennan P, Munafo M, Pring M, et al. A multivariable mendelian randomization analysis investigating smoking and alcohol consumption in oral and oropharyngeal cancer. Nat Commun. 2020;11:6071.
Li S, Chen H, Man J, Zhang T, Yin X, He Q, Yang X, Lu M. Changing trends in the disease burden of esophageal cancer in China from 1990 to 2017 and its predicted level in 25 years. Cancer Med. 2021;10:1889–99.
Cumberbatch MGK, Jubber I, Black PC, Esperto F, Figueroa JD, Kamat AM, Kiemeney L, Lotan Y, Pang K, Silverman DT, et al. Epidemiology of bladder Cancer: a systematic review and contemporary update of risk factors in 2018. Eur Urol. 2018;74:784–95.
Praud D, Rota M, Pelucchi C, Bertuccio P, Rosso T, Galeone C, Zhang ZF, Matsuo K, Ito H, Hu J, et al. Cigarette smoking and gastric cancer in the stomach Cancer Pooling (StoP) project. Eur J Cancer Prev. 2018;27:124–33.
Yi M, Li A, Zhou L, Chu Q, Song Y, Wu K. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: estimates based on the global burden of disease study 2017. J Hematol Oncol. 2020;13:72.
Chen P, Liu Y, Wen Y, Zhou C. Non-small cell lung cancer in China. Cancer Commun (Lond). 2022;42:937–70.
Warren GW, Cummings KM. Tobacco and lung cancer: risks, trends, and outcomes in patients with cancer. Am Soc Clin Oncol Educ Book 2013:359–64.
Zhu J, Wang J, Wang T, Zhou H, Xu M, Zha J, Feng C, Shen Z, Jiang Y, Chen J. Identification of molecular subtypes, risk signature, and immune landscape mediated by necroptosis-related genes in non-small cell lung cancer. Front Oncol. 2022;12:955186.
Yang C, Wei Y, Li W, Wei J, Chen G, Xu M, Liu G. Prognostic Risk Signature and Comprehensive Analyses of Endoplasmic Reticulum Stress-Related Genes in Lung Adenocarcinoma. J Immunol Res. 2022, 2022:6567916.
Chen DT, Hsu YL, Fulp WJ, Coppola D, Haura EB, Yeatman TJ, Cress WD. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J Natl Cancer Inst. 2011;103:1859–70.
Giannos P, Kechagias KS, Gal A. Identification of prognostic gene biomarkers in Non-small Cell Lung Cancer Progression by Integrated Bioinformatics Analysis. Biology (Basel) 2021, 10.
Yang D, Liu Y, Bai C, Wang X, Powell CA. Epidemiology of lung cancer and lung cancer screening programs in China and the United States. Cancer Lett. 2020;468:82–7.
Park CK, Cho HJ, Choi YD, Oh IJ, Kim YC. A phase II trial of Osimertinib in the second-line treatment of non-small cell Lung Cancer with the EGFR T790M mutation, detected from circulating Tumor DNA: LiquidLung-O-Cohort 2. Cancer Res Treat. 2019;51:777–87.
Zheng H, Zhan Y, Liu S, Lu J, Luo J, Feng J, Fan S. The roles of tumor-derived exosomes in non-small cell lung cancer and their clinical implications. J Exp Clin Cancer Res. 2018;37:226.
Cassim S, Chepulis L, Keenan R, Kidd J, Firth M, Lawrenson R. Patient and carer perceived barriers to early presentation and diagnosis of lung cancer: a systematic review. BMC Cancer. 2019;19:25.
Ding H, Shi L, Chen Z, Lu Y, Tian Z, **ao H, Deng X, Chen P, Zhang Y. Construction and evaluation of a prognostic risk model of tumor metastasis-related genes in patients with non-small cell lung cancer. BMC Med Genomics. 2022;15:187.
Balata H, Fong KM, Hendriks LE, Lam S, Ostroff JS, Peled N, Wu N, Aggarwal C. Prevention and early detection for NSCLC: advances in thoracic oncology 2018. J Thorac Oncol. 2019;14:1513–27.
Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat Res Rev Mutat Res. 2021;787:108365.
Steuer CE, Jegede OA, Dahlberg SE, Wakelee HA, Keller SM, Tester WJ, Gandara DR, Graziano SL, Adjei AA, Butts CA, et al. Smoking behavior in patients with early-stage NSCLC: a Report from ECOG-ACRIN 1505 Trial. J Thorac Oncol. 2021;16:960–7.
Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34.
Guo W, Hu M, Wu J, Zhou A, Liao Y, Song H, Xu D, Kuang Y, Wang T, **g B, et al. Gprc5a depletion enhances the risk of smoking-induced lung tumorigenesis and mortality. Biomed Pharmacother. 2019;114:108791.
Zhang MY, Liu XX, Li H, Li R, Liu X, Qu YQ. Elevated mRNA levels of AURKA, CDC20 and TPX2 are associated with poor prognosis of smoking related lung adenocarcinoma using bioinformatics analysis. Int J Med Sci. 2018;15:1676–85.
Wu C, Liu T, Shi Z, Fang L, ** H, Tong P. Novel nomograms for predicting the risk of low distal bone strength: development and validation in a Chinese population-based observational study. J Orthop Surg Res. 2023;18:70.
Balachandran VP, Gonen M, Smith JJ, DeMatteo RP. Nomograms in oncology: more than meets the eye. Lancet Oncol. 2015;16:e173–180.
Mandrekar JN. Receiver operating characteristic curve in diagnostic test assessment. J Thorac Oncol. 2010;5:1315–6.
Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. 2017;127:4042–58.
Sibille A, Corhay JL, Louis R, Ninane V, Jerusalem G, Duysinx B. Eosinophils and Lung Cancer: from bench to Bedside. Int J Mol Sci 2022, 23.
Crinier A, Narni-Mancinelli E, Ugolini S, Vivier E. SnapShot: natural killer cells. Cell. 2020;180:1280–e12801281.
Crotty S. T Follicular Helper Cell Biology: a decade of Discovery and diseases. Immunity. 2019;50:1132–48.
Chen Y, Jiang T, Zhang H, Gou X, Han C, Wang J, Chen AT, Ma J, Liu J, Chen Z, et al. LRRC31 inhibits DNA repair and sensitizes breast cancer brain metastasis to radiation therapy. Nat Cell Biol. 2020;22:1276–85.
Yao S, Xu J, Zhao K, Song P, Yan Q, Fan W, Li W, Lu C. Down-regulation of HPGD by miR-146b-3p promotes cervical cancer cell proliferation, migration and anchorage-independent growth through activation of STAT3 and AKT pathways. Cell Death Dis. 2018;9:1055.
Zhang J, Rong J, Ge W, Wang J, Wang W, Chi H. SPINK5 is a Tumor-Suppressor Gene Involved in the Progression of Nonsmall Cell Lung Carcinoma through Negatively Regulating PSIP1. J Healthc Eng. 2022, 2022:2209979.
Cai H, Jiao Y, Li Y, Yang Z, He M, Liu Y. Low CYP24A1 mRNA expression and its role in prognosis of breast cancer. Sci Rep. 2019;9:13714.
Hsu YL, Hung JY, Liang YY, Lin YS, Tsai MJ, Chou SH, Lu CY, Kuo PL. S100P interacts with integrin alpha7 and increases cancer cell migration and invasion in lung cancer. Oncotarget. 2015;6:29585–98.
Zhang X, Wang F, Huang Y, Ke K, Zhao B, Chen L, Liao N, Wang L, Li Q, Liu X, et al. FGG promotes migration and invasion in hepatocellular carcinoma cells through activating epithelial to mesenchymal transition. Cancer Manag Res. 2019;11:1653–65.
Zhou C, Chen H, Han L, Xue F, Wang A, Liang YJ. Screening of genes related to lung cancer caused by smoking with RNA-Seq. Eur Rev Med Pharmacol Sci. 2014;18:117–25.
Liu Q, Niu X, Li Y, Zhang JR, Zhu SJ, Yang QY, Zhang W, Gong L. Role of the mucin-like glycoprotein FCGBP in mucosal immunity and cancer. Front Immunol. 2022;13:863317.
Acknowledgements
Some elements of the Fig. 10 are from BioRender.com.
Funding
This work was supported by the “Six-one” Project for High-level Health Talents (LGY2016037), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX21-3108), Nantong Key Laboratory of Translational Medicine in Cardiothoracic Diseases, Nantong Clinical Medical Research Center of Cardiothoracic Disease, and Institution of Translational Medicine in Cardiothoracic Diseases in Affiliated Hospital of Nantong University. National Natural Science Foundation of China (82370253), Jiangsu Provincial Research Hospital (YJXYY202204), Innovation Team Project of Affiliated Hospital of Nantong University (XNBHCX31773).
Author information
Authors and Affiliations
Contributions
QL and TW contributed equally to this work. Study concept and design: JS and YZ; Data collection and processing: QL, YT, ZT and XZ; Technical and material support: QL, TW and ZS; Experiment implementation and date analysis: QL, TW and YT; Paper writing: QL and TW; Funding support and study supervision: TW, JS and YZ. All authors contributed to the study and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.



12935_2024_3347_MOESM3_ESM.jpg
Supplementary Material 3: Fig. S3 Mutation and survival analysis of FCGBP in NSCLC. (A) Mutation frequency of seven prognostic smoking-related genes in 1121 NSCLC samples. (B) Expression level of FCGBP in NSCLC smoker and non-smoking NSCLC patients. (C) The prognosis of NSCLC smokers with low expression of FCGBP is significantly worse than that of smokers with high expression of FCGBP
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Li, Q., Wang, T., Tang, Y. et al. A novel prognostic signature based on smoking-associated genes for predicting prognosis and immune microenvironment in NSCLC smokers. Cancer Cell Int 24, 171 (2024). https://doi.org/10.1186/s12935-024-03347-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03347-9