
Abstract
The tumor microenvironment is overwhelmingly dictated by macrophages, intimately affiliated with tumors, exercising pivotal roles in multiple processes, including angiogenesis, extracellular matrix reconfiguration, cellular proliferation, metastasis, and immunosuppression. They further exhibit resilience to chemotherapy and immunotherapy via meticulous checkpoint blockades. When appropriately stimulated, macrophages can morph into a potent bidirectional component of the immune system, engulfing malignant cells and annihilating them with cytotoxic substances, thus rendering them intriguing candidates for therapeutic targets. As myelomonocytic cells relentlessly amass within tumor tissues, macrophages rise as prime contenders for cell therapy upon the development of chimeric antigen receptor effector cells. Given the significant incidence of macrophage infiltration correlated with an unfavorable prognosis and heightened resistance to chemotherapy in solid tumors, we delve into the intricate role of macrophages in cancer propagation and their promising potential in confronting four formidable cancer variants—namely, melanoma, colon, glioma, and breast cancers.
Similar content being viewed by others
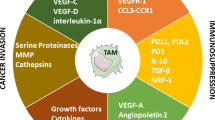
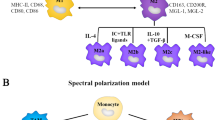
Introduction
Cancer immunotherapy has become a potent disease treatment option that helps advanced cancer patients survive longer while removing any chance of returning tumors [1, 2]. In cancer patients, immune cells are ineffective against cancer cells and promote tumor growth, decreasing treatment effectiveness [3, 4]. Among innate system cells, macrophages play a crucial role in normal homeostasis, inflammation, and phagocytosis [5, 6]. However, macrophages have been shown to play a negative role in the progression of oncogenesis and neoplastic disease by promoting genetic instability and angiogenesis. [7]. Macrophages are divided into the M1 and M2 subgroups based on morphological, phenotypic, and functional variability. The M2 macrophages have been shown to support tumor growth and metastasis, whereas the M1 macrophages play a crucial role in antitumor immunity and largely orchestrate pro-inflammatory activities in the tumor microenvironment (TME) [8, 9]. Tumor-associated macrophages (TAMs), the most diversified immune cells in the TME that are essential for tumor formation, include the M2 macrophages and a small population of M1 macrophages [10]. In this line, tumor cells secrete chemokines and growth factors to draw in macrophages and change them into the M2 type. Therefore, it was also discovered that significant dynamic changes in macrophage subpopulations were related to the efficacy of immunotherapy [30]. Moreover, the secretion of STAT3 by TAMs into the TME, with their increasing numbers in the stroma, can lead to CD8+ T cell exhaustion [31]. TAMs and myeloid-derived suppressor cells can also suppress immune function through cell-to-cell contact, stimulating myeloid-derived suppressor cells (MDSCs) to secrete IL-10 and inhibit IL-12 production via dendritic cells [32]. The TAMs also play a role in inhibiting T cell recruitment, so targeting certain pathways, such as colony-stimulating factor-1 (CSF1/CSF1R) [3], can obstruct macrophage recruitment and promote T cell infiltration [33].
TAMs targeting breast cancer therapy
Currently, CSF-1R is inhibited by PLX3397 to diminish M2 macrophage recruitment, which is utilized to treat malignancies such as glioblastoma, breast cancer, and other tumors. There was high tolerability in a phase 1 study of the CSF-1R inhibitor LY3022855 in metastatic breast cancer [34]. Twenty-two medicines that target CXCR4 are now in the active development phase; most of these are small molecule antagonists; however, there are also antibody-based medications, gene therapies, and CAR-T cell treatments. Eighteen of these medications are being developed to treat solid tumors and hematological malignancies. Mozobil (Plerixafo), a small molecule antagonist that targets CXCR4, was introduced in 2018. It is first utilized with granulocyte colony-stimulating factor (G-CSF) to provoke hematopoietic stem cells for therapy of multiple myeloma and non-lymphoma Hodgkin’s.
To evaluate IMM2902’s safety and effectiveness in HER2+ advanced solid tumors, clinical trials have approved the drug's primary indication of the lung, gastric HER2-positive breast, and other solid tumors (NCT05076591). SIGLEC10 interacts with CD24 in renal clear cell carcinoma, triple-negative breast, and ovarian cancers to prevent tumor cell phagocytosis and immune cell activation. Blocking SIGLEC10hi TAMs in HCC decreased the expression of immunosuppressive molecules and increased the cytotoxic effects of CD8+ T cells. It also supported Pembrolizumab as an anti-tumor drug that targets PD-1 molecules [74]. Wei, C. et al. showed that IL-6 secretion by TAMs stimulates EMT, thereby improving CRC invasion and migration ability through the modulation of the JAK2/STAT3/miR-506-3p/FoxQ1 axis [80, 81]. For instance, in a 30-patient Japanese CRC cohort, a lower density of CD68+ in the tumor stroma and invasive front were linked to more progressive cancer, whereas high levels of TAMs were linked to a favorable prognosis [82]. Similar relationships got observed in European cohorts. For instance, in a tissue microarray of 100 colon cancer patients in Germany demonstrated decreased CD68+ macrophages in higher-stage tumors [83]. In a Bulgarian cohort conducted on 210 patients with primary CRC, a lower density of CD68+ TAMs in the invasive tumor front which is considerably associated with the advanced tumor stage (III and IV stages), distant metastases, and local lymph nodes specific metastases was observed [84]. A lesser number of CD68+ TAMs were also reported in cancer patients where the tumor cells migrated and invaded the blood circulation, lymph vessels, and perineural tissues.
Additionally, a high CD206/CD68 ratio has been linked to improved recurrence-free survival rates in patients with stage II of CRC after receiving adjuvant chemotherapy [85]. On the other hand, both VEGF-expressing and CD68-TAMs have been found to predict improved survival rates in individuals with stages II and III of CRC. So, only TAM infiltration cannot fully explain the degree of disease recurrence. A recent meta-analysis of 6115 CRC cases from 27 separate studies indicated a high density of TAMs in CRC as an independent favorable predictor for 5-year OS but not for DFS. The TAM density and additional prognostic markers may be a more accurate predictor of CRC relapse. In this line, traditional methods of analyzing TAMs have relied on the expression of CD68, a pan-macrophage marker, but recent studies have used double immunofluorescence staining to identify different subsets of TAMs using other markers such as CD86, CD163, and CD206 [86, 87]. Literature has shown that the presence of M2 macrophages (CD163+) is correlated with poorer overall survival and disease-free survival/recurrence-free survival in CRC. In addition, high levels of CD163+ TAMs and a high CD163/CD68 ratio have been linked to an aggressive phenotype and poor prognosis in CRC [80]. Additionally, the expression of CD86 TAMs and TNM stage were found to be independent prognostic factors for recurrence-free survival and overall survival in CRC [88].
Potential applications of TAMs in CRC therapy
Blocking monocyte infiltration in CRC
Blocking the infiltration of mononuclear cells, such as TAMs, in the inflammatory tissues associated with tumors has been identified as a potential therapeutic method for primary cancers. Chanmee et al. demonstrated that TAMs, specifically those associated with colon cancer, induce CXCR4, CXCL-12, and HIF-1 in the hypoxic TME. Moreover, the accumulation of TAMs is blocked by targeting the HIF-1/CXCR4 axis effectively. Mantovani et al. revealed that TAMs derived from colon cancer monocytes could differentiate, highlighting the need for combination therapies that block differentiation to target these cells effectively. In this line, the TNF-α has been found to induce the recruitment of monocytes and simultaneously inhibit the differentiation of monocytes or macrophages into TAMs in the TME of colon cancer in vivo [89]. Another strategy for targeting TAMs is the inhibition of their recruitment or infiltration. SIX1, a protein that is overexpressed in various types of cancer and promotes the recruitment of pro-tumor TAMs to the region of colorectal cancer (CRC) [79], can be silenced through the use of its inhibitor, Nitazoxanide, which suppresses the WNT/CTNNB1 pathway [90, 91]. Trifluridine/Tipiracil, an anti-metabolism drug, has been observed to effectively exhaust M2 macrophages when combined with oxaliplatin, leading to the infiltration of cytotoxic CD8+ T cells and the lysis of tumor cells [92].
Repolarizing TAMs
TAMs predominantly exhibit an M2 phenotype, simultaneously promoting immunosuppression and angiogenesis. They can be re-educated via M2 to M1 polarization. For instance, TAMs mediated inhibition of macrophage receptor expression with collagenous structure (MARCO) repolarized TAMs to the M1 phenotype and caused antitumor activity in the MC38 colon cancer mice model [93]. By altering the number and frequency of myeloid cells infiltrating the tumor, tasquinimod-based immunotherapy can reduce the immunosuppressive potential of TME [94]. It has been demonstrated by Olsson et al. that tasquinimod targets early-stage myeloid cells that tend to penetrate tumors, causing M2 myeloid cells to adopt an M1 macrophage phenotype, altering the tumor microenvironment, preventing angiogenesis, and inhibiting metastatic spread [95].CRC can be diagnosed and treated using long non-coding RNAs (lncRNA) as noninvasive biomarkers and targeted molecules. For instance, cells secreting lncRNAs, such as RPPH1, promote M2 polarization and tumor metastasis but can't be directly targeted [90, 91]. Cathepsin K (CTSK), which binds to TLR4 and activates mTOR, is synthesized by intestinal microflora and modulates the expression of long noncoding RNAs in various tissues [92]. The CTSK-specific inhibitor Odanacatib has been reported to curb the pro-tumor effects and improve the prognosis of CRC patients [96]. Moreover, researchers found that Ru@ICG-BLZ nanoparticles effectively repolarize TAMs to M1 macrophages due to their CRC specificity and low toxic properties as a new approach [97].
Targeting TAMs in immunotherapy
Immune checkpoint inhibitors, T cells-based treatment, and autologous tumor vaccines are the key components of immunotherapy in CRC treatment [98,99,100,101]. These strategies target immune checkpoint inhibitors with matching targets, including CTLA-4, PD-1, and PD-L1 [3, 10, 102]. The co-inhibitory molecule CTLA-4, produced by T cells, binds to the ligand CD80/86 on adenomatous polyposis coli (APC) to produce an inhibitory signal [103]. When PD-1, an immunosuppressive receptor on T cells, binds to PD-L1, it significantly reduces the activity of antigen-specific T cells [104]. PD-L1 is expressed mainly by aggressive primary tumor cells and by CD68/CD163-positive M2 macrophages in patients with colorectal cancer with high microsatellite instability [105]. Gordon and his team discovered that when the illness worsens, TAMs express more PD-1. Additional research revealed the ability of TAMs to phagocytose is inversely linked with PD-1 expression, and in vivo, inhibition of PD-1-PD-L1 improved macrophage phagocytosis, slowed tumor growth, and lengthened mouse survival [16]. On the other hand, evidence suggests that the PD-1/PD-L1 axis plays a role in skewing TAMs from the M1 to M2 phenotype, and M2 TAMs have been found to contribute to resistance to PD-1/PD-L1 blockade [133, 134]. Repolarizing TAMs by immunomodulators such as imiquimod and IFNs may inhibit melanoma tumor growth as TAMs generated by CCL17 and CCL22 attract Tregs to melanoma tumor locations [135, 136]. Certain chemokines, including IL-8, CCL4, CCL17, and CXCL10, in the cerebrospinal fluid may predict brain metastases in melanoma patients. The use of certain chemotherapy agents, such as nimustine hydrochloride, dacarbazine, and vincristine, has been shown to decrease CCL22 production in B16F10 melanoma mice [134]. These findings suggest that TAM-derived chemokines produced in the tumor stroma under the influence of POSTN (a protein involved in extracellular matrix organization) may contribute to melanoma-specific TILs in melanoma patients [137].
Targeting TAMs-derived angiogenic factors
TAMs have been shown to produce various angiogenic factors, including platelet-derived growth factor (PDGF), VEGF, TGFβ, and matrix metalloproteinases (MMPs) [138]. These factors can promote neovascularization by recruiting TAMs to the location of skin tumors in mouse models [139]. The VEGF and MMPs have been identified as crucial indicators of skin cancer progression [140], with high concentrations of POSTN and CD163+ TAMs in the tumor stroma of skin malignancies leading to increased production of MMP1 and MMP12 in skin lesions [141]. TAMs stimulated by tumor stromal factors may therefore serve as potential targets for molecular targeted therapy in the treatment of cancers [133].
Effects of anti-cancer agents on TAMs
Recent studies have also concentrated on the immunomodulatory impacts of chemotherapeutic drugs on TAMs. For instance, in mouse melanoma models, non-cytotoxic dosages of paclitaxel could reduce MDSCs and even prevent their ability to suppress the immune response [142]. It has been discovered that chemotherapeutic agents and drugs with low molecular weight co-localize along with TAMs at tumor locations. Hu-Lieskovan et al. showed combination therapy with dabrafenib and trametinib with synergistic impacts of immune checkpoint inhibitors. In contrast, dabrafenib and trametinib monotherapy led to elevated Tregs and decreased TAMs in melanoma, respectively [143].
In a different study, the collagen-structured anti-macrophage receptor was discovered to induce TAMs polarization into pro-inflammatory phenotypes, leading to anti-tumor immunological responses in B16 melanomas [36]. Furthermore, Gordon et al. showed that suppression of PD-1/PD-L1 in vivo promoted macrophage phagocytosis, decreased tumor progression, and improved macrophage survival [16, 144]. Lymphocyte-Activation Gene 3 (LAG-3, CD223) is another important immune checkpoint molecule that participates in T-cell exhaustion similar to Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) and Programmed cell death protein 1 (PD-1) [145]. It is expressed on the surface of activated CD4+ and CD8+ T cells and other immune cells, such as natural killer cells, regulatory T cells, and macrophages [146, 147]. TAMs release chemokines that lead to the recruitment of immune-suppressive cells towards the tumor microenvironment, which influence other stromal cells, like fibroblasts, to synthesize chemokines. Young et al. demonstrated that granulocytic MDSCs are recruited to tumor sites by the CXCR2 ligand, produced by fibroblasts, after being stimulated by IL-1β from TAMs [148]. Moreover, combination therapy with anti-CD115 Abs and CXCR2 agonists might inhibit B16F10 melanoma in vivo by preventing the enrollment of granulocytic MDSCs and removing immature TAMs [149]. Notably, emactuzumab, an anti-human CD115 Ab, reduced CD163+ CD206+ M2 macrophages in melanoma cases by eliminating immature TAMs before being stimulated by IL-4 [150]. These findings imply that anti-CXCR2 agonists and emactuzumab may trigger the anti-melanoma immune response by lowering M2 polarized TAMs. These results highlight the necessity of understanding how chemotherapeutic agents affect TAMs. The ICIs, in combination with TAMs targeting agents, provide promising outcomes for melanoma treatment. Data from preclinical research provided good explanations for clinical trials in which elimination or repolarization of immunosuppressive TAMs are being investigated to overcome ICI resistance and improve their anti-tumor functions [151]. Studies using ICIs and immunomodulatory factors that block M2-TAMs activities have been performed or are still being conducted in melanoma patients. Decreases in M-CSF (CSF-1) and increases in GM-CSF levels are two strategies being investigated in conjunction with ICIs to re-polarize M2-TAMs into M1-TAMs. For example, the phase 2 studies of recombinant human analog (sargramostim) as a GM-CSF agonist in combination with ipilimumab for the treatment of unresectable stage III or IV metastatic melanoma has been completed and revealed increased survival [152,153,154]. Talimogene laherparepvec (T-VEC), a modified oncolytic herpes virus, is another GM-CSF agonist that increases the anti-tumor responses and has been approved for local treatment of advanced melanoma. The T-VEC exclusively infects and replicates in tumor cells and results in immune-mediated lysis of tumor cells via encoding human GM-CSF, as well as the susceptibility of melanoma to ICIs. Combination therapy of melanoma with T-VEC plus nivolumab and pembrolizumab has reached phase 2 clinical trial [155,156,157]. OPTiM is also a phase III trial of talimogene laherparepvec, in which T-VEC had long-term efficacy in contrast to GM-CSF in advanced melanoma [158, 159]. Moreover, ONCOS-102 is an engineered oncolytic adenovirus encoding GM-CSF that has shown synergistic effects for metastatic or unrespectable melanoma treatment in combination with pembrolizumab (anti-PD-1 Ab) [160, 161]. Designing antagonists against M-CSF cytokine is another strategy that might lead to the depletion of M2-TAMs and an improvement in ICIs functions. In addition, M-CSF contributes to metastatic melanoma resistance to BRAF-targeted therapies. Therefore, M-CSF acts as a therapeutic target in BRAFV600E melanoma. Monoclonal antibody lacnotuzumab, an anti-M-CSF, has been studied alone and in combination with ICI spartalizumab (an anti-PD-1 mAb) [162,163,164,165]. The M-CSF receptor (CSF1R) provides another therapeutic target to deplete the immunosuppressive functions of TAMs. Some examples include BLZ945 (CSF1R inhibitor) combined with PDR001 (anti-PD-1 mAb), LY3022855 (CSF1R inhibitor) combined with tremelimumab or durvalumab ICIs, emactuzumab (CSF1R inhibitor), and cabiralizumab (a humanized mAb against CSF1R) [166,167,168,169,170]. APX005M is a humanized CD40 agonist mAb that activates immune responses by stimulating IFN-γ secretion [170]. INCB001158 is an arginase inhibitor used as monotherapy or combined with pembrolizumab in solid metastatic tumors such as melanoma. It has been suggested that inhibition of metabolic enzymes, such as ARG-1, could restore T-cell activities by filling arginine storage [171]. Moreover, it has been reported that PI3K-γ inhibition can re-polarize M2-TAMs into pro-inflammatory M1-TAMs. Moreover, IPI-549 is a PI3K inhibitor used alone or in combination with nivolumab (Fig. 5) [172, 173].

Role of TAMs in melanoma occurrence and therapy. Melanoma cells can elicit an immune response through the release of various cytokines, including CXCL-2, CCL-2, CSF-1, GM-CSF, A9, S100A8, and VEGFA. Some of these cytokines, such as GM-CSF and VEGFA, can stimulate the activation of macrophages, transforming these cells into TAMs. The activation of macrophages also results in the release of a series of factors, including TGF-β, CCL-22, and IL-10, which can influence dendritic cells and T-lymphocytes. In addition, TAMs can release TNF-α and interferon-γ to target cancer cells. It is worth noting that matrix metalloproteinases 9 and 2 (MMP9 and MMP2) can break down collagen in the tissue surrounding the melanoma mass, contributing to its decomposition
Macrophages in glioma cancer (GBM)
Glioma is a type of primary brain tumor, including glioblastoma, astrocytoma, and oligodendroglioma [174]. The microenvironment of glioma is characterized by the presence of macrophages and microglia, known as tumor-associated macrophages [175, 176]. Microglia, which are phagocytes of the central nervous system, exist in three forms: amoeboid, ramified, and reactive [177, 178]. Amoeboid microglia are involved in embryonic central nervous system development [179], while ramified microglia are found in large quantities in the brain parenchyma with the ability to transform into neurons, astrocytes, or oligodendrocytes [180, 181]. Reactive microglia, which are rod-like with non-branching processes and numerous lysosomes and phagosomes, are associated with brain injury and neuroinflammation [182, 183]. They also secrete MHC class II antigens and produce inflammatory mediators [184, 185]. Macrophages in the central nervous system can be classified according to their location as perivascular macrophages, meningeal macrophages, macrophages of the circumventricular organs, or macrophages of the choroid plexus. Among the brain cancers mentioned above, glioma is a particularly aggressive and untreatable type of brain tumor with a poor prognosis, and current treatments have not been successful in improving outcomes [186, 187]. Therefore, there is a need for further research into the mechanisms behind the invasiveness and recurrence of glioma and the development of new therapeutic approaches, including immunologic treatment [188, 189].
Glioma-associated macrophages (GAMs) are a key component of the tumor microenvironment in gliomas [190] that can be derived from microglia as well as bone marrow-derived macrophages [191,192,193,194]. The number and characteristics of GAMs can vary significantly, with evidence from single-cell sequencing showing that GAMs are made up of 59.05% and 27.87% of immunocytes in primary and recurrent glioblastomas, respectively [195]. Various signaling molecules, growth factors, transcription factors, and epigenetic and post-transcriptional modifications influence the phenotype and activation state of GAMs. Depending on their origin, these cells can exhibit different characteristics, with some derived from brain-resident microglia [196] and others from bone marrow-derived monocytes [197]. The GAMs play a role in various aspects of glioma progression, such as cell motility, proliferation, survival [188], and immune suppression [198, 199]. They can also produce a range of growth factors and pro-inflammatory cytokines that contribute to the supportive matrix for tumor cell metastasis and the development of an immunosuppressive microenvironment [200]. Understanding the role of GAMs in the tumor microenvironment may provide insights into potential therapeutic approaches for gliomas. In this context, Woolf et al. demonstrated using single-cell imaging that P2RY12 and TMEM119 label microglia in GBM, and they further demonstrated that these markers could be used to distinguish microglia from BMDM. P2RY12 protein expression is associated with longer survival rates in patients. Activation of P2Y12 receptors has been linked to the extension of microglial cell processes [201, 202]. Moreover, another study that analyzed marker genes in GAMs found that only a small number of genes were consistently present, indicating the diverse responses observed in different settings. In this regard, Tgm2 and Gpnmb genes were the only ones that were common across the analyzed data sets, highlighting the need for further research to understand the functional state of GAMs.
GAMs regulating GBM malignancy
In the presence of glioblastoma (GBM) cells, the functions of microglia may be impaired, leading to the initiation or growth of tumors. This has been demonstrated through comparative transcriptome analysis. It was found that GBM-bearing mice's microglia are less sensitive and impaired at monitoring immunity due to a reduction in a group of genes that encode receptors for various antigens, chemokines, and cytokines [203]. Additionally, microglia engage in reciprocal molecular crosstalk with glioblastoma stem cells, exhibiting a more direct pro-tumorigenic function through the secretion of TGF-β [204]. Microglia activated by GM-CSF can release CCL5, a chemokine that upregulates the secretion of MMP2 in GBM cells, thereby promoting tumor migration and invasion [205]. This effect may be mediated by the secretion of interferon-gamma (IFNγ) by infiltrating microglia, which leads to the stable expression of a specific transcriptional program in GBM cells that is associated with myeloid cells [206]. This epigenetic immunoediting may also be present in human mesenchymal subtype glioblastoma stem cells (GSCs) [207]. The TAMs also play a role in GBM invasion through the expression of CCL8 and the activation of signaling pathways in GBM cells through the binding of CCL8 to CCR1 and CCR5 receptors [208], the secretion of CSF-1 [209] and epidermal growth factor (EGF) by GBM and microglia, respectively, have also been shown to stimulate GBM invasion through the recruitment of TAMs and activation of signaling pathways in GBM cells through the binding of EGF to epidermal growth factor receptors (EGFR) [210].
GAMs in angiogenesis of GBM
The resistance of GBM to anti-VEGF therapy, which targets a protein involved in angiogenesis, has been linked to the macrophages infiltration into the tumor (Fig. 6) [211]. It depends on the activation state of the immune cells and whether they promote or suppress angiogenesis. Immunosuppressive macrophages like M2 promote angiogenesis, while pro-inflammatory macrophages like M1 suppress it [212]. Depletion of TAMs in animal models has been shown to reduce the blood vessel density in GBM, suggesting a role of these cells in GBM angiogenesis [213]. Resident microglia may be particularly important in this process, as their selective depletion has been shown to reduce blood vessels in GBM to a greater extent than the depletion of all TAMs [213]. The TAMs isolated from a specific type of glioma have been found to overexpress proangiogenic factors such as VEGF and CXCL2, both of which have been linked to angiogenesis. The interaction of the receptor for advanced glycation end products (RAGE) with its ligands has also been shown to promote angiogenesis in GBM through the activation of TAMs-specific signaling pathways [214].

Anti-tumor/pro-tumor activity of macrophages in GBM
GAMs in drug resistance of GBM
Resistance to temozolomide (TMZ) has been reported as a common obstacle to GBM patients’ treatment, where the resistance rate is approximately 60% [215]. Literature evidence suggests that genetic factors and GAMs may contribute to this resistance [216]. The interleukin-11 (IL-11) produced by microglia and macrophages activates STAT3-MYC signaling in GBM cells, leading to TMZ resistance [216]. By inhibiting GAM recruitment and IL-11 secretion through ABP1 ablation or genetic inactivation, TMZ resistance has been reversed in a murine model of GBM [217]. Additionally, different subpopulations of GAMs may have distinct effects on treatment responses [218]. For instance, M2-like GAMs contribute to resistance through secretion of exosomal miR-21-5p, while M1-like polarization of GAMs induced by GBM-derived extracellular HMGB1 has been shown to restore sensitivity to TMZ. In addition to chemoresistance, GAMs have also been implicated in resistance to radiotherapy and antiangiogenic therapy [219]. The impact of GAMs on treatment responses may be mediated by the expression of PD-L1, which interacts with CD80 on T-cells and leads to CD4+ T-cell suppression, Treg expansion, and immune checkpoint blockade resistance [220]. The role of CD73-expressing macrophages in ICB resistance has also been demonstrated in a murine model of GBM [221].
GAM-targeted therapy in GBM
Several approaches have been identified and tested in experimental and clinical settings for targeting TAMs in glioblastoma (GBM). These approaches can be divided into three categories: TAM re-education, TAM education, and TAM depletion. TAM education involves activating pro-inflammatory pathways, which can also be delivered through gene therapy or direct administration, while TAM depletion involves targeting key molecules to achieve the unbiased depletion of TAMs or to inhibit macrophage infiltration. These TAM-targeting strategies can potentially counter immunotherapies and influence glioma progression [174].
Anti-angiogenic treatment
Tumor-infiltrating myeloid cells may play a role in the limited effectiveness of anti-angiogenic therapies by expressing alternative proangiogenic factors that bypass VEGF-mediated pathways [222]. The MerTK inhibitor MRX-2843 has been shown to have therapeutic benefits by promoting the polarization of macrophages away from immunosuppressive conditions, inhibiting neo-angiogenesis in the glioblastoma microenvironment, and inducing tumor cell death [223]. The metalloprotease-disintegrin ADAM8, which is highly expressed in tumor cells and associated immune cells in glioblastomas, is related to angiogenesis and is associated with a poor clinical prognosis [224]. The regulation of osteopontin mediates the angiogenic potential of ADAM8 in glioblastoma cells/primary macrophages, so targeting ADAM8 may be a viable approach for modulating angiogenesis in glioblastoma [225].
PD-L1 signaling pathway
Pembrolizumab monotherapy, which targets the PD-1 protein, cannot elicit an effective immune response in most GBM patients, likely due to the low number of T cells within the tumor microenvironment and the abundance of CD68 + macrophages [226]. Besides that, in a recent study, it was reported that GBM cells secrete interleukin-11 (IL11) in response to glial-derived neurotrophic GAMs, activating signal transducer and activator of transcription 3-MYC signaling. This signaling pathway leads to the induction of stem cell states, which increase tumorigenicity and resistance to temozolomide (TMZ) in GBM cells. In mouse GBM models, PI3K inactivation or inhibition reduces microglia recruitment and IL11 secretion, resulting in improved TMZ response [227]. Anyway, in the late stage of temozolomide (TMZ) treatment or relapse, treatment with an anti-PD-L1 antibody significantly reduced the infiltration of CD163-positive macrophages into tumors. In contrast, a combination of a PD-L1 antibody and IPI-549 (a selective PI3Kγ inhibitor) therapy effectively inhibited tumor growth [228]. Treatment with rapamycin and hydroxychloroquine (RQ) decreased the polarization of M2 macrophages, increased phagocytic ability, and increased the accumulation of lipid droplets. This treatment enhanced the ratio of anti-tumoral to pro-tumoral immune cells within the tumor and the ratio of CD8 to CD4 T cells. The combination of RQ and anti-PD1 treatment was found to be synergistic in action [229]. Saha et al. tested a triple combination of anti-CTLA-4, anti-PD-1, and G47Δ-mIL12 (oncolytic herpes simplex viruses armed with angiostatin and IL-12) in mouse GBM models. This treatment was associated with an influx of macrophages, an anti-tumoral, macrophage-like polarization of these cells, and an increase in the ratio of T-effector to T regulatory cells. This treatment was able to cure most mice with gliomas. Immune cell depletion studies showed that CD4+ and CD8+ T cells and macrophages are all required for the synergistic curative activity of this treatment [230].
Combination therapy
Several studies have reported the potential of targeting pro-tumoral macrophages in the treatment of GBM. Almahariq et al. found that the BLZ-945, a CSF-1R inhibitor, reduced pro-tumoral macrophage polarization and improved the response to radiotherapy in respected tumors with a high baseline population of pro-tumoral macrophages [231]. The results of Zhu et al. showed that when debulking plus anti-CD47 tumors were compared with non-debulking plus IgG tumors, macrophages with CD68-positive labels were recruited more, pro-inflammatory cytokines like CXCL10 were increased, and angiogenic proteins were decreased, indicating that surgical resections coupled with anti-CD47 blocking immunotherapy promote inflammation and prolong survival [232]. As a result of lipopolysaccharide and interferon-gamma stimulation of bone marrow macrophages and brain-resident macrophages, Herting et al. have found that dexamethasone prevented the production of IL-1. These findings suggest that IL-1 signaling may be a useful therapeutic target in the management of GBM-associated cerebral edema [233] (Table 1).
Conclusion
The use of cancer immunotherapy for removing residual tumors has emerged as an effective way to improve the survival of patients with advanced-stage cancers, as it enhances the immune system's ability to eliminate minimal residual tumors. As a result of ineffective immune cells against cancer cells, patients with cancer are more likely to develop tumors, which reduces the effectiveness of therapeutic measures. The macrophage is one of the most important innate system cells contributing to normal homeostasis, inflammation, and phagocytosis. Several studies have shown, however, that macrophages promote genetic instability and angiogenesis in the development of oncogenesis and neoplasms. The M2 macrophages promote tumor growth and metastasis. Among the most diverse immune cells in the TME are the M2 macrophages, which along with the M1 macrophages, are called TAMs. The pro-tumorigenic M2 macrophages are attracted to tumor cells by chemokines and growth factors. Therefore, immunotherapy efficacy is also strongly influenced by changes in macrophage subpopulations. The TAMs have been implicated as a therapeutic target in numerous biological studies due to their ability to deplete, inhibit recruitment, and influence polarization status. In addition, TAMs limit the efficacy of immunotherapy approaches, such as anti-PD1 treatment, because they are linked to resistance to well-known antitumor therapies, such as chemotherapy and radiotherapy. Anyway, many preclinical studies using small molecules or antibodies to block each of mentioned factors/pathways individually have demonstrated significant improvement in response to a wide variety of tumors to therapy, indicating that their blockage is generally well tolerated. However, more research is needed to overcome macrophage-based cancer therapy, particularly in nanoparticles and drug delivery. In this line, the use of small molecules or antibodies to block specific factors or pathways associated with TAMs has shown promising results in preclinical studies, leading to improved responses to a wide variety of tumors. These approaches have generally been well tolerated. However, more research is needed, especially in the field of nanoparticles and drug delivery, to advance macrophage-based cancer therapy further. As the role of TAMs in cancer therapy is increasingly recognized, several crucial gaps in the field necessitate further investigation. TAM heterogeneity, plasticity, and their interactions with other immune cells remain areas of exploration. Understanding the underlying mechanisms of TAM-mediated immunosuppression and identifying reliable biomarkers for patient stratification and treatment response assessment is paramount. Additionally, optimizing TAM-targeted therapies and validating their clinical effectiveness are essential for translating preclinical findings into meaningful treatments.
Availability of data and materials
Not applicable.
Abbreviations
- TME:
-
Tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- IBC:
-
Inflammatory breast cancer
- DCIS:
-
Ductal carcinoma in situ
- IDC:
-
Invasive ductal carcinoma
- CSCs:
-
Cancer stem cells
- EMT:
-
Epithelial–mesenchymal transition
- MDSCs:
-
Myeloid-derived suppressor cells
- CSF1/CSF1R:
-
Colony stimulating factor 1
- Gpr132:
-
G Protein-Coupled Receptor 132
- HIFs:
-
Hypoxia-inducible factors
- TNFα:
-
Tumor necrosis factor-alpha
- TGFβ1:
-
Transforming growth factor beta 1
- SIGLEC1:
-
Sialic acid-binding Ig-like lectin 1
- G-CSF:
-
Granulocyte colony-stimulating factor
- MCP1:
-
Monocyte chemotactic protein-1
- MIP-1 and MIP-2:
-
Macrophage inflammatory proteins-1 and -2
- MK2:
-
MAPK-activated protein kinase 2
- CRC:
-
Colorectal cancer
- lncRNA:
-
Long non-coding RNAs
- TICs:
-
Tumor-initiating cells
- MTF:
-
Tumor cell fusion
- Tregs:
-
Regulatory T cells
- MDSCs:
-
Myeloid-derived suppressor cells
- MARCO:
-
Macrophage receptor with collagenous structure
- GBM:
-
Glioblastoma
References
Kennedy LB, Salama AK. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104.
Razi S, Haghparast A, Chodari Khameneh S, Ebrahimi Sadrabadi A, Aziziyan F, Bakhtiyari M, Nabi-Afjadi M, Tarhriz V, Jalili A, Zalpoor H. The role of tumor microenvironment on cancer stem cell fate in solid tumors. Cell Commun Signal. 2023;21(1):1–23.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82.
Mohammadi N, Fayazi Hosseini N, Nemati H, Moradi-Sardareh H, Nabi-Afjadi M, Kardar GA. Revisiting of properties and modified polyethylenimine-based cancer gene delivery systems. Biochem Genet. 2023. https://doi.org/10.1007/s10528-023-10416-7.
DiPietro LA, Wilgus TA, Koh TJ. Macrophages in healing wounds: paradoxes and paradigms. Int J Mol Sci. 2021;22(2):950.
Zalpoor H, Aziziyan F, Liaghat M, Bakhtiyari M, Akbari A, Nabi-Afjadi M, Forghaniesfidvajani R, Rezaei N. The roles of metabolic profiles and intracellular signaling pathways of tumor microenvironment cells in angiogenesis of solid tumors. Cell Commun Signal. 2022;20(1):186.
Mantovani A, Ponzetta A, Inforzato A, Jaillon S. Innate immunity, inflammation and tumour progression: double-edged swords. J Intern Med. 2019;285:524–32.
Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37.
Khesht AMS, Karpisheh V, Saeed BQ, Zekiy AO, Yapanto LM, Afjadi MN, Aksoun M, Esfahani MN, Aghakhani F, Movahed M. Different T cell related immunological profiles in COVID-19 patients compared to healthy controls. Int Immunopharmacol. 2021;97: 107828.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416.
**ong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, Nickles D, Cubas R. Anti–PD-L1 Treatment results in functional remodeling of the macrophage compartment anti–PD-L1 treatment can remodel the macrophage compartment. Can Res. 2019;79(7):1493–506.
Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, Ji X, Liu W, Huang B, Luo W. Breast cancer development and progression: risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5(2):77–106.
Takeya M, Komohara Y. Role of tumor-associated macrophages in human malignancies: friend or foe? Pathol Int. 2016;66(9):491–505.
Yang Q, Guo N, Zhou Y, Chen J, Wei Q, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B. 2020;10(11):2156–70.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Investig. 2012;122(3):787–95.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9.
Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174(5):1293-1308.e1236.
Chen X, Yu T, Zhang J, Li Y, Chen H, Yang G, Yu W, Liu Y, Liu X, Duan C. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene. 2017;36(35):5045–57.
Liu Q, Hodge J, Wang J, Wang Y, Wang L, Singh UP, Li Y, Yao Y, Wang D, Ai W. Emodin reduces breast cancer lung metastasis by suppressing macrophage-induced breast cancer cell epithelial–mesenchymal transition and cancer stem cell formation. Theranostics. 2020;10(18):8365.
Guo L, Cheng X, Chen H, Chen C, **e S, Zhao M, Liu D, Deng Q, Liu Y, Wang X. Induction of breast cancer stem cells by M1 macrophages through Lin-28B-let-7-HMGA2 axis. Cancer Lett. 2019;452:213–25.
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, Zhong H, Schneider RJ. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network chemokines and macrophages in inflammatory breast cancer. Can Res. 2019;79(13):3360–71.
Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi M, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin A. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31(11):1354–65.
Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schübeler D, van Nimwegen E, Christofori G. Sox4 is a master regulator of epithelial–mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23(6):768–83.
Choi J, Gyamfi J, Jang H, Koo JS. The role of tumor-associated macrophage in breast cancer biology. Histol Histopathol. 2018. https://doi.org/10.14670/HH-11-916.
Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26(1):1–13.
de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. Oncoimmunology. 2012;1(8):1305–12.
Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M, Giannini M, Cognigni PD, Ferrara G, Battelli N. Triple negative breast cancer: Key role of tumor-associated macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim et Biophys Acta BBA Rev Cancer. 2018;1869(1):78–84.
Joshi N, Hajizadeh F, Dezfouli EA, Zekiy AO, Afjadi MN, Mousavi SM, Hojjat-Farsangi M, Karpisheh V, Mahmoodpoor A, Hassannia H. Silencing STAT3 enhances sensitivity of cancer cells to doxorubicin and inhibits tumor progression. Life Sci. 2021;275: 119369.
Hossain MA, Liu G, Dai B, Si Y, Yang Q, Wazir J, Birnbaumer L, Yang Y. Reinvigorating exhausted CD8+ cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med Res Rev. 2021;41(1):156–201.
Xu L, Li C. Single-cell transcriptome analysis reveals the M2 macrophages and exhausted T cells and intratumoral heterogeneity in triple-negative breast cancer. Anti-Cancer Agents Med Chem (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 2022;22(2):294–312.
Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509–21.
Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Investig. 2015;125(9):3365–76.
Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55(7-8–9):861–7.
Autio KA, Klebanoff CA, Schaer D, Kauh JS, Slovin SF, Blinder VS, Comen EA, Danila DC, Hoffman DM, Kang S. Phase 1 study of LY3022855, a colony-stimulating factor-1 receptor (CSF-1R) inhibitor, in patients with metastatic breast cancer (MBC) or metastatic castration-resistant prostate cancer (MCRPC). American Society of Clinical Oncology; 2019.
**ao N, Zhu X, Li K, Chen Y, Liu X, Xu B, Lei M, Xu J, Sun H-C. Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol. 2021;10(1):1–14.
Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, Dahan R, Harris RA, Rantalainen M, Klevebring D. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15(9):2000–11.
Esmaily M, Masjedi A, Hallaj S, Afjadi MN, Malakotikhah F, Ghani S, Ahmadi A, Sojoodi M, Hassannia H, Atyabi F. Blockade of CTLA-4 increases anti-tumor response inducing potential of dendritic cell vaccine. J Control Release. 2020;326:63–74.
Yu T, Di G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin J Cancer Res. 2017;29(3):237.
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med. 2008;205(6):1261–8.
Vickers NJ. Animal communication: when i’m calling you, will you answer too? Curr Biol. 2017;27(14):R713–5.
Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat. 2014;148(2):291–302.
Wang S, Wang N, Huang X, Yang B, Zheng Y, Zhang J, Wang X, Lin Y, Wang Z. Baohuoside i suppresses breast cancer metastasis by downregulating the tumor-associated macrophages/CXC motif chemokine ligand 1 pathway. Phytomedicine. 2020;78: 153331.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Ogino S, Nowak JA, Hamada T, Phipps AI, Peters U, Milner DA Jr, Giovannucci EL, Nishihara R, Giannakis M, Garrett WS. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut. 2018;67(6):1168–80.
Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–39.
Alexander PG, McMillan DC, Park JH. The local inflammatory response in colorectal cancer–type, location or density? A systematic review and meta-analysis. Cancer Treat Rev. 2020;83: 101949.
Shalapour S, Karin M. Pas de deux: control of anti-tumor immunity by cancer-associated inflammation. Immunity. 2019;51(1):15–26.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Kwak Y, Koh J, Kim DW, Kang SB, Kim WH, Lee HS. Immunoscore encompassing CD3+ and CD8+ T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget. 2016;7(49):81778–90.
Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472–9.
Zhou Q, Peng RQ, Wu XJ, **a Q, Hou JH, Ding Y, Zhou QM, Zhang X, Pang ZZ, Wan DS, et al. The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J Transl Med. 2010;8:13.
Grizzi F, Bianchi P, Malesci A, Laghi L. Prognostic value of innate and adaptive immunity in colorectal cancer. World J Gastroenterol WJG. 2013;19(2):174.
Li S, Xu F, Zhang J, Wang L, Zheng Y, Wu X, Wang J, Huang Q, Lai M. Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. Oncoimmunology. 2018;7(2): e1380765.
Ohnishi K, Komohara Y, Saito Y, Miyamoto Y, Watanabe M, Baba H, Takeya M. CD 169-positive macrophages in regional lymph nodes are associated with a favorable prognosis in patients with colorectal carcinoma. Cancer Sci. 2013;104(9):1237–44.
Kim Y, Wen X, Bae JM, Kim JH, Cho NY, Kang GH. The distribution of intratumoral macrophages correlates with molecular phenotypes and impacts prognosis in colorectal carcinoma. Histopathology. 2018;73(4):663–71.
Algars A, Irjala H, Vaittinen S, Huhtinen H, Sundström J, Salmi M, Ristamäki R, Jalkanen S. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. Int J Cancer. 2012;131(4):864–73.
Kawada M, Seno H, Kanda K, Nakanishi Y, Akitake R, Komekado H, Kawada K, Sakai Y, Mizoguchi E, Chiba T. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene. 2012;31(26):3111–23.
Zhu C, Chrifi I, Mustafa D, van der Weiden M, Leenen PJM, Duncker DJ, Kros JM, Cheng C. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene. 2017;36(38):5356–68.
Huang JK, Ma L, Song WH, Lu BY, Huang YB, Dong HM, Ma XK, Zhu ZZ, Zhou R. LncRNA-MALAT1 promotes angiogenesis of thyroid cancer by modulating tumor-associated macrophage FGF2 protein secretion. J Cell Biochem. 2017;118(12):4821–30.
Zhu C, Kros JM, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol. 2017;19(11):1435–46.
Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10(1):58.
Badawi MA, Abouelfadl DM, El-Sharkawy SL, El-Aal WE, Abbas NF. Tumor-associated macrophage (TAM) and angiogenesis in human colon carcinoma. Open Access Macedonian J Med Sci. 2015;3(2):209–14.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008;27(1):11–8.
Hotchkiss KA, Ashton AW, Klein RS, Lenzi ML, Zhu GH, Schwartz EL. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Can Res. 2003;63(2):527–33.
Suarez-Lopez L, Kong YW, Sriram G, Patterson JC, Rosenberg S, Morandell S, Haigis KM, Yaffe MB. MAPKAP Kinase-2 drives expression of angiogenic factors by tumor-associated macrophages in a model of inflammation-induced colon cancer. Front Immunol. 2020;11: 607891.
Luput L, Licarete E, Sesarman A, Patras L, Alupei MC, Banciu M. Tumor-associated macrophages favor C26 murine colon carcinoma cell proliferation in an oxidative stress-dependent manner. Oncol Rep. 2017;37(4):2472–80.
Vinnakota K, Zhang Y, Selvanesan BC, Topi G, Salim T, Sand-Dejmek J, Jönsson G, Sjölander A. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J Cell Physiol. 2017;232(12):3468–80.
Cassetta L, Pollard JW. Repolarizing macrophages improves breast cancer therapy. Cell Res. 2017;27(8):963–4.
Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R, Severgnini M, De Bellis G, Sidney J, Sette A, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut. 2017;66(3):454–63.
Zhao P, Wang B, Zhang Z, Zhang W, Liu Y. Response gene to complement 32 expression in macrophages augments paracrine stimulation-mediated colon cancer progression. Cell Death Dis. 2019;10(10):776.
Yu X, Wang D, Wang X, Sun S, Zhang Y, Wang S, Miao R, Xu X, Qu X. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J Exp Clin Cancer Res CR. 2019;38(1):32.
Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene. 2016;35(44):5735–45.
Phinney BB, Ray AL, Peretti AS, Jerman SJ, Grim C, Pinchuk IV, Beswick EJ. MK2 regulates macrophage chemokine activity and recruitment to promote colon tumor growth. Front Immunol. 1857;2018:9.
Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, Liu Q, Dou R, **ong B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019;18(1):64.
Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102.
Yang C, Wei C, Wang S, Shi D, Zhang C, Lin X, Dou R, **ong B. Elevated CD163(+)/CD68(+) ratio at tumor invasive front is closely associated with aggressive phenotype and poor prognosis in colorectal cancer. Int J Biol Sci. 2019;15(5):984–98.
Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019;79(1):146–58.
Xu H, Zhang Y, Peña MM, Pirisi L, Creek KE. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis. 2017;38(3):281–92.
Koelzer VH, Canonica K, Dawson H, Sokol L, Karamitopoulou-Diamantis E, Lugli A, Zlobec I. Phenoty** of tumor-associated macrophages in colorectal cancer: impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology. 2016;5(4): e1106677.
Li J, Li L, Li Y, Long Y, Zhao Q, Ouyang Y, Bao W, Gong K. Tumor-associated macrophage infiltration and prognosis in colorectal cancer: systematic review and meta-analysis. Int J Colorectal Dis. 2020;35(7):1203–10.
Nakayama Y, Nagashima N, Minagawa N, Inoue Y, Katsuki T, Onitsuka K, Sako T, Hirata K, Nagata N, Itoh H. Relationships between tumor-associated macrophages and clinicopathological factors in patients with colorectal cancer. Anticancer Res. 2002;22(6c):4291–6.
Sickert D, Aust DE, Langer S, Haupt I, Baretton GB, Dieter P. Characterization of macrophage subpopulations in colon cancer using tissue microarrays. Histopathology. 2005;46(5):515–21.
Gulubova M, Ananiev J, Yovchev Y, Julianov A, Karashmalakov A, Vlaykova T. The density of macrophages in colorectal cancer is inversely correlated to TGF-β1 expression and patients’ survival. J Mol Histol. 2013;44(6):679–92.
Feng Q, Chang W, Mao Y, He G, Zheng P, Tang W, Wei Y, Ren L, Zhu D, Ji M, et al. Tumor-associated macrophages as prognostic and predictive biomarkers for postoperative adjuvant chemotherapy in patients with stage II colon cancer. Clin Cancer Res. 2019;25(13):3896–907.
Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180–5.
Zhao Y, Ge X, Xu X, Yu S, Wang J, Sun L. Prognostic value and clinicopathological roles of phenotypes of tumour-associated macrophages in colorectal cancer. J Cancer Res Clin Oncol. 2019;145(12):3005–19.
Kou Y, Li Z, Sun Q, Yang S, Wang Y, Hu C, Gu H, Wang H, Xu H, Li Y, et al. Prognostic value and predictive biomarkers of phenotypes of tumour-associated macrophages in colorectal cancer. Scand J Immunol. 2022;95(4): e13137.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55.
Qu Y, Olsen JR, Yuan X, Cheng PF, Levesque MP, Brokstad KA, Hoffman PS, Oyan AM, Zhang W, Kalland KH, et al. Small molecule promotes β-catenin citrullination and inhibits Wnt signaling in cancer. Nat Chem Biol. 2018;14(1):94–101.
Song W, Ma J, Lei B, Yuan X, Cheng B, Yang H, Wang M, Feng Z, Wang L. Sine oculis homeobox 1 promotes proliferation and migration of human colorectal cancer cells through activation of Wnt/β-catenin signaling. Cancer Sci. 2019;110(2):608–16.
Limagne E, Thibaudin M, Nuttin L, Spill A, Derangère V, Fumet JD, Amellal N, Peranzoni E, Cattan V, Ghiringhelli F. Trifluridine/tipiracil plus oxaliplatin improves PD-1 blockade in colorectal cancer by inducing immunogenic cell death and depleting macrophages. Cancer Immunol Res. 2019;7(12):1958–69.
Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6(3):1670–90.
Shen L, Sundstedt A, Ciesielski M, Miles KM, Celander M, Adelaiye R, Orillion A, Ciamporcero E, Ramakrishnan S, Ellis L. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine modelstasquinimod enhances cancer immunotherapies. Cancer Immunol Res. 2015;3(2):136–48.
Olsson A, Nakhlé J, Sundstedt A, Plas P, Bauchet A-L, Pierron V, Bruetschy L, Deronic A, Törngren M, Liberg D. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J Immunother Cancer. 2015;3(1):1–14.
Pacheco-Fernández T, Juárez-Avelar I, Illescas O, Terrazas LI, Hernández-Pando R, Pérez-Plasencia C, Gutiérrez-Cirlos EB, Ávila-Moreno F, Chirino YI, Reyes JL, et al. Macrophage migration inhibitory factor promotes the interaction between the tumor, macrophages, and T cells to regulate the progression of chemically induced colitis-associated colorectal cancer. Mediators Inflamm. 2019;2019:2056085.
Zalpoor H, Bakhtiyari M, Liaghat M, Nabi-Afjadi M, Ganjalikhani-Hakemi M. Quercetin potential effects against SARS-CoV-2 infection and COVID-19-associated cancer progression by inhibiting mTOR and hypoxia-inducible factor-1α (HIF-1α). Phytother Res. 2022. https://doi.org/10.1002/ptr.7440.
Matos AI, Carreira B, Peres C, Moura LI, Conniot J, Fourniols T, Scomparin A, Martínez-Barriocanal Á, Arango D, Conde JP. Nanotechnology is an important strategy for combinational innovative chemo-immunotherapies against colorectal cancer. J Control Release. 2019;307:108–38.
Feng M, Zhao Z, Yang M, Ji J, Zhu D. T-cell-based immunotherapy in colorectal cancer. Cancer Lett. 2021;498:201–9.
Johdi NA, Sukor NF. Colorectal cancer immunotherapy: options and strategies. Front Immunol. 2020;11:1624.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16(6):361–75.
Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11(1):1–26.
Beyranvand Nejad E, van der Sluis TC, van Duikeren S, Yagita H, Janssen GM, van Veelen PA, Melief CJ, van der Burg SH, Arens R. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Can Res. 2016;76(20):6017–29.
Cortese N, Soldani C, Franceschini B, Barbagallo M, Marchesi F, Torzilli G, Donadon M. Macrophages in colorectal cancer liver metastases. Cancers. 2019;11(5):633.
Korehisa S, Oki E, Iimori M, Nakaji Y, Shimokawa M, Saeki H, Okano S, Oda Y, Maehara Y. Clinical significance of programmed cell death-ligand 1 expression and the immune microenvironment at the invasive front of colorectal cancers with high microsatellite instability. Int J Cancer. 2018;142(4):822–32.
Wei Y, Liang M, **ong L, Su N, Gao X, Jiang Z. PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp Cell Res. 2021;402(2): 112575.
Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, Bompas E, Piperno-Neumann S, Cousin S, Grellety T, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. 2018;4(1):93–7.
Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206–21.
Li W, Wu F, Zhao S, Shi P, Wang S, Cui D. Correlation between PD-1/PD-L1 expression and polarization in tumor-associated macrophages: a key player in tumor immunotherapy. Cytokine Growth Factor Rev. 2022;67:49–57.
Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, Minutolo NG, Casado-Medrano V, Lopez G, Matsuyama T, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. 2021;12(1):877.
Tacconi C, Ungaro F, Correale C, Arena V, Massimino L, Detmar M, Spinelli A, Carvello M, Mazzone M, Oliveira AI, et al. Activation of the VEGFC/VEGFR3 pathway induces tumor immune escape in colorectal cancer. Cancer Res. 2019;79(16):4196–210.
Massi D, Marconi C, Franchi A, Bianchini F, Paglierani M, Ketabchi S, Miracco C, Santucci M, Calorini L. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: evidence from human and experimental tumors. Hum Pathol. 2007;38(10):1516–25.
Bianchini F, Massi D, Marconi C, Franchi A, Baroni G, Santucci M, Mannini A, Mugnai G, Calorini L. Expression of cyclo-oxygenase-2 in macrophages associated with cutaneous melanoma at different stages of progression. Prostaglandins Other Lipid Mediat. 2007;83(4):320–8.
Kale S, Raja R, Thorat D, Soundararajan G, Patil T, Kundu G. Erratum: Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via α9β1 integrin. Oncogene. 2015;34(42):5408–10.
Hollander L, Guo X, Velazquez H, Chang J, Safirstein R, Kluger H, Cha C, Desir GV. Renalase expression by melanoma and tumor-associated macrophages promotes tumor growth through a STAT3-mediated mechanismtreatment of melanoma using inhibitors of renalase signaling. Can Res. 2016;76(13):3884–94.
Kim O-H, Kang G-H, Noh H, Cha J-Y, Lee H-J, Yoon J-H, Mamura M, Nam J-S, Lee DH, Kim YA. Proangiogenic TIE2 (+)/CD31 (+) macrophages are the predominant population of tumor-associated macrophages infiltrating metastatic lymph nodes (vol 36, pg 432, 2013); 2014.
Huber R, Meier B, Otsuka A, Fenini G, Satoh T, Gehrke S, Widmer D, Levesque MP, Mangana J, Kerl K. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci Rep. 2016;6(1):1–14.
Tham M, Tan KW, Keeble J, Wang X, Hubert S, Barron L, Tan NS, Kato M, Prevost-Blondel A, Angeli V. Melanoma-initiating cells exploit M2 macrophage TGFβ and arginase pathway for survival and proliferation. Oncotarget. 2014;5(23):12027.
Clawson GA, Matters GL, **n P, Imamura-Kawasawa Y, Du Z, Thiboutot DM, Helm KF, Neves RI, Abraham T. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS ONE. 2015;10(8): e0134320.
Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, Brys L, Abels C, Lahmar Q, Ergen C. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Can Res. 2016;76(1):35–42.
Huang SC-C, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. 2016;45(4):817–30.
Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198(3):1006–14.
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–42.
Klimp A, De Vries E, Scherphof G, Daemen T. A potential role of macrophage activation in the treatment of cancer. Crit Rev Oncol Hematol. 2002;44(2):143–61.
Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, Lewis KD, Tawbi HA, Hernandez G, Wongchenko MJ. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med. 2019;25(6):929–35.
Gogas H, Dréno B, Larkin J, Demidov L, Stroyakovskiy D, Eroglu Z, Ferrucci PF, Pigozzo J, Rutkowski P, Mackiewicz J. Cobimetinib plus atezolizumab in BRAFV600 wild-type melanoma: primary results from the randomized phase III IMspire170 study. Ann Oncol. 2021;32(3):384–94.
Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P, Demidov L. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2020;395(10240):1835–44.
Dummer R, Queirolo P, Guijarro AMA, Hu Y, Wang D, de Azevedo SJ, Robert C, Ascierto PA, Chiarion-Sileni V, Pronzato P. Atezolizumab, vemurafenib, and cobimetinib in patients with melanoma with CNS metastases (TRICOTEL): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2022;23(9):1145–55.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–46.
Wang H, Zhang L, Yang L, Liu C, Zhang Q, Zhang L. Targeting macrophage anti-tumor activity to suppress melanoma progression. Oncotarget. 2017;8(11):18486.
Lesinski GB. The potential for targeting the STAT3 pathway as a novel therapy for melanoma. Future Oncol. 2013;9(7):925–7.
Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L, Yu X, Luo Q, Zhang Z. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11(9):9536–49.
Furudate S, Fujimura T, Kakizaki A, Kambayashi Y, Asano M, Watabe A, Aiba S. The possible interaction between periostin expressed by cancer stroma and tumor-associated macrophages in develo** mycosis fungoides. Exp Dermatol. 2016;25(2):107–12.
Fujimura T, Kakizaki A, Kambayashi Y, Sato Y, Tanita K, Lyu C, Furudate S, Aiba S. Cytotoxic antimelanoma drugs suppress the activation of M2 macrophages. Exp Dermatol. 2018;27(1):64–70.
Kakizaki A, Fujimura T, Furudate S, Kambayashi Y, Yamauchi T, Yagita H, Aiba S. Immunomodulatory effect of peritumorally administered interferon-beta on melanoma through tumor-associated macrophages. Oncoimmunology. 2015;4(11): e1047584.
Furudate S, Fujimura T, Kambayashi Y, Kakizaki A, Hidaka T, Aiba S. Immunomodulatory effect of imiquimod through CCL22 produced by tumor-associated macrophages in B16F10 melanomas. Anticancer Res. 2017;37(7):3461–71.
Lok E, Chung AS, Swanson KD, Wong ET. Melanoma brain metastasis globally reconfigures chemokine and cytokine profiles in patient cerebrospinal fluid. Melanoma Res. 2014;24(2):120.
Linde N, Lederle W, Depner S, Van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol. 2012;227(1):17–28.
Zhu X, Yang J, Gao Y, Wu C, Yi L, Li G, Qi Y. The dual effects of a novel peptibody on angiogenesis inhibition and M2 macrophage polarization on sarcoma. Cancer Lett. 2018;416:1–10.
Yamada K, Uchiyama A, Uehara A, Perera B, Ogino S, Yokoyama Y, Takeuchi Y, Udey MC, Ishikawa O. Motegi S-i: MFG-E8 drives melanoma growth by stimulating mesenchymal stromal cell-induced angiogenesis and M2 polarization of tumor-associated macrophagesMFG-E8 promotes MSC-induced angiogenesis and TAM in melanoma. Can Res. 2016;76(14):4283–92.
Kale S, Raja R, Thorat D, Soundararajan G, Patil T, Kundu G. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via α9β1 integrin. Oncogene. 2014;33(18):2295–306.
Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, Shurin GV, Shurin MR, Umansky V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol. 2013;190(5):2464–71.
Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF V600E melanoma. Sci Transl Med. 2015;7(279):279ra241-279ra241.
Sefid F, Payandeh Z, Azamirad G, Baradaran B, Nabi Afjadi M, Islami M, Darvish M, Kalantar SM, Kahroba H, Ardakani MA. Atezolizumab and granzyme B as immunotoxin against PD-L1 antigen; an insilico study. In Silico Pharmacol. 2021;9(1):1–12.
Chocarro L, Bocanegra A, Blanco E, Fernández-Rubio L, Arasanz H, Echaide M, Garnica M, Ramos P, Piñeiro-Hermida S, Vera R. Cutting-edge: preclinical and clinical development of the first approved Lag-3 inhibitor. Cells. 2022;11(15):2351.
Gertel S, Polachek A, Elkayam O, Furer V. Lymphocyte activation gene-3 (LAG-3) regulatory T cells: an evolving biomarker for treatment response in autoimmune diseases. Autoimmun Rev. 2022;21(6): 103085.
Zhao L, Wang H, Xu K, Liu X, He Y. Update on lymphocyte-activation gene 3 (LAG-3) in cancers: from biological properties to clinical applications. Chin Med J. 2022;135(10):1203–12.
Young HL, Rowling EJ, Bugatti M, Giurisato E, Luheshi N, Arozarena I, Acosta J-C, Kamarashev J, Frederick DT, Cooper ZA. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J Exp Med. 2017;214(6):1691–710.
Lyons YA, Pradeep S, Wu SY, Haemmerle M, Hansen JM, Wagner MJ, Villar-Prados A, Nagaraja AS, Dood RL, Previs RA. Macrophage depletion through colony stimulating factor 1 receptor pathway blockade overcomes adaptive resistance to anti-VEGF therapy. Oncotarget. 2017;8(57):96496.
Pradel LP, Ooi C-H, Romagnoli S, Cannarile MA, Sade H, Rüttinger D, Ries CH. Macrophage susceptibility to emactuzumab (RG7155) treatment. Mol Cancer Ther. 2016;15(12):3077–86.
Anfray C, Ummarino A, Torres Andon F, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. 2019;9(1):46.
Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, Leming P, Puzanov I, Shin D, Kirkwood JM. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744–53.
Cham J, Zhang L, Kwek S, Paciorek A, He T, Fong G, Oh DY, Fong L. Combination immunotherapy induces distinct T-cell repertoire responses when administered to patients with different malignancies. J Immunother Cancer. 2020;8(1):e000368.
Kwek SS, Kahn J, Greaney SK, Lewis J, Cha E, Zhang L, Weber RW, Leonard L, Markovic SN, Fong L. GM-CSF and ipilimumab therapy in metastatic melanoma: clinical outcomes and immunologic responses. Oncoimmunology. 2016;5(4): e1101204.
Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers. 2021;13(6):1383.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Goins WF, Huang S, Hall B, Marzulli M, Cohen JB, Glorioso JC. Engineering HSV-1 vectors for gene therapy. In: Herpes Simplex Virus: Methods and Protocols 2014;63–79.
Andtbacka RH, Collichio F, Harrington KJ, Middleton MR, Downey G, Ӧhrling K, Kaufman HL. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J Immunother Cancer. 2019;7(1):1–11.
Andtbacka RH, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, Amatruda T, Zager JS, Cranmer L, Hsueh E. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol. 2016;23(13):4169–77.
Kuryk L. Møller A-SW, Jaderberg M: Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology. 2019;8(2): e1532763.
Shoushtari AN, Olszanski AJ, Nyakas M, Hornyak TJ, Wolchok JD, Levitsky V, Kuryk L, Hansen TB, Jäderberg M. Pilot study of ONCOS-102 and pembrolizumab: remodeling of the tumor microenvironment and clinical outcomes in anti–PD-1–resistant advanced melanoma. Clin Cancer Res. 2022;29:1–10.
Barceló C, Sisó P, de la Rosa I, Megino-Luque C, Navaridas R, Maiques O, Urdanibia I, Eritja N, Soria X, Potrony M. M-CSF as a therapeutic target in BRAFV600E melanoma resistant to BRAF inhibitors. Br J Cancer. 2022;127(6):1142–52.
Pognan F, Couttet P, Demin I, Jaitner B, Pang Y, Roubenoff R, Sutter E, Timsit Y, Valentin MA, Vogel B. Colony-stimulating factor-1 antibody lacnotuzumab in a phase 1 healthy volunteer study and mechanistic investigation of safety outcomes. J Pharmacol Exp Ther. 2019;369(3):428–42.
Wu H, Zhou X, Wang X, Cheng W, Hu X, Wang Y, Luo B, Huang W, Gu J. miR-34a in extracellular vesicles from bone marrow mesenchymal stem cells reduces rheumatoid arthritis inflammation via the cyclin I/ATM/ATR/p53 axis. J Cell Mol Med. 2021;25(4):1896–910.
Calvo A, Joensuu H, Sebastian M, Naing A, Bang Y-J, Martin M, Roda D, Hodi FS, Veloso A, Mataraza J. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. American Society of Clinical Oncology; 2018.
Lin C-C, Gil-Martin M, Bauer TM, Naing A. Lim DW-T, Sarantopoulos J, Geva R, Ando Y, Fan L, Choudhury S: Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Can Res. 2020;80(16_supplement):CT171.
Falchook GS, Peeters M, Rottey S, Dirix LY, Obermannova R, Cohen JE, Perets R, Frommer RS, Bauer TM, Wang JS. A phase 1a/1b trial of CSF-1R inhibitor LY3022855 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. Invest New Drugs. 2021;39(5):1284–97.
Dowlati A, Harvey RD, Carvajal RD, Hamid O, Klempner SJ, Kauh JSW, Peterson DA, Yu D, Chapman SC, Szpurka AM. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: phase 1 dose-escalation trial. Invest New Drugs. 2021;39(4):1057–71.
Gomez-Roca C, Cassier P, Zamarin D, Machiels J-P, Gracia JLP, Hodi FS, Taus A, Garcia MM, Boni V, Eder JP. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J Immunother Cancer. 2022;10(5):e004076.
Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, Ralabate A, Johnson B, Levit NS, Anderson G. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1Phase I study of APX005M, cabiralizumab, and nivolumab. Clin Cancer Res. 2021;27(17):4757–67.
Koyama T, Shimizu T, Matsubara N, Iwasa S, Naito Y, Kondo S, Harano K, Yonemori K, Kotani D, Yoh K. MO10-6 Phase 1 study of retifanlimab (anti-PD-1) and INCB001158 (arginase inhibitor), alone or in combination, in solid tumors. Ann Oncol. 2021;32:S302.
Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, Ribas A, Brail LH, Roberts J, Lee L. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. American Society of Clinical Oncology; 2018.
Postow M, Sullivan R, Cohen E, Gutierrez M, Hong D, Steuer C, McCarter J, Zizlsperger N, Kutok J, O’Connell B. 434 Updated clinical data from the melanoma expansion cohort of an ongoing Ph1/1b Study of eganelisib (formerly IPI-549) in combination with nivolumab. BMJ Specialist Journals; 2020.
Andersen JK, Miletic H, Hossain JA. Tumor-associated macrophages in gliomas—basic insights and treatment opportunities. Cancers. 2022;14(5):1319.
Zhou W, Bao S. Reciprocal supportive interplay between glioblastoma and tumor-associated macrophages. Cancers. 2014;6(2):723–40.
Hughes-Austin JM, Deane KD, Derber LA, Kolfenbach JR, Zerbe GO, Sokolove J, Lahey LJ, Weisman MH, Buckner JH, Mikuls TR, et al. Multiple cytokines and chemokines are associated with rheumatoid arthritis-related autoimmunity in first-degree relatives without rheumatoid arthritis: Studies of the Aetiology of Rheumatoid Arthritis (SERA). Ann Rheum Dis. 2013;72(6):901–7.
Prionisti I, Bühler LH, Walker PR, Jolivet RB. Harnessing microglia and macrophages for the treatment of glioblastoma. Front Pharmacol. 2019;10:506.
Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–70.
Fetler L, Amigorena S. Brain under surveillance: the microglia patrol. Science. 2005;309(5733):392–3.
Ling E, Penney D, Leblond C. Use of carbon labeling to demonstrate the role of blood monocytes as precursors of the ‘ameboid cells’ present in the corpus callosum of postnatal rats. J Comp Neurol. 1980;193(3):631–57.
Dalmau I, Finsen B, Zimmer J, González B, Castellano B. Development of microglia in the postnatal rat hippocampus. Hippocampus. 1998;8(5):458–74.
Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33(3):256–66.
Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–8.
Benveniste EN, Nguyen VT, O’Keefe GM. Immunological aspects of microglia: relevance to Alzheimer’s disease. Neurochem Int. 2001;39(5–6):381–91.
Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. J Immunol. 1998;160(10):4671–80.
Wang H, Xu T, Huang Q, ** W, Chen J. Immunotherapy for malignant glioma: current status and future directions. Trends Pharmacol Sci. 2020;41(2):123–38.
Phillips RE, Soshnev AA, Allis CD. Epigenomic reprogramming as a driver of malignant glioma. Cancer Cell. 2020;38(5):647–60.
Nejo T, Yamamichi A, Almeida ND, Goretsky YE, Okada H. Tumor antigens in glioma. In: Seminars in immunology. Elsevier; 2020. p. 101385.
Hervey-Jumper SL, Berger MS. Insular glioma surgery: an evolution of thought and practice: JNSPG 75th Anniversary Invited Review Article. J Neurosurg. 2019;130(1):9–16.
Radin DP, Tsirka SE. Interactions between tumor cells, neurons, and microglia in the glioma microenvironment. Int J Mol Sci. 2020;21(22):8476.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441.
Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J Neuroinflamm. 2017;14(1):1–11.
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–7.
Arcuri C, Fioretti B, Bianchi R, Mecca C, Tubaro C, Beccari T, Franciolini F, Giambanco I, Donato R. Microglia-glioma cross-talk: a two way approach to new strategies against glioma. Front Biosci Landmark. 2017;22(2):268–309.
Fu W, Wang W, Li H, Jiao Y, Huo R, Yan Z, Wang J, Wang S, Wang J, Chen D. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front Immunol. 2020;11:835.
Xu C, **ao M, Li X, **n L, Song J, Zhan Q, Wang C, Zhang Q, Yuan X, Tan Y. Origin, activation, and targeted therapy of glioma-associated macrophages. Front Immunol. 2022;13:974996.
Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, Holtman IR, Wang X, Eggen BJ, Boddeke HW. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE. 2015;10(2): e0116644.
Wei J, Gabrusiewicz K, Heimberger A. The controversial role of microglia in malignant gliomas. Clin Dev Immunol. 2013;2013:285246.
McFarland BC, Marks MP, Rowse AL, Fehling SC, Gerigk M, Qin H, Benveniste EN. Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget. 2016;7(15):20621.
Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S, Haller J, Yan RE, Ziober A, Nguyen A. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun. 2019;7(1):1–12.
Saavedra-López E, Roig-Martínez M, Cribaro GP, Casanova PV, Gallego JM, Pérez-Vallés A, Barcia C. Phagocytic glioblastoma-associated microglia and macrophages populate invading pseudopalisades. Brain commun. 2020;2(1):fcz043.
Milior G, Morin-Brureau M, Chali F, Le Duigou C, Savary E, Huberfeld G, Rouach N, Pallud J, Capelle L, Navarro V. Distinct P2Y receptors mediate extension and retraction of microglial processes in epileptic and peritumoral human tissue. J Neurosci. 2020;40(7):1373–88.
Maas SL, Abels ER, Van De Haar LL, Zhang X, Morsett L, Sil S, Guedes J, Sen P, Prabhakar S, Hickman SE. Glioblastoma hijacks microglial gene expression to support tumor growth. J Neuroinflamm. 2020;17(1):1–18.
Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J Mol Med. 2018;42(6):3395–403.
Yu-Ju WuC, Chen C-H, Lin C-Y, Feng L-Y, Lin Y-C, Wei K-C, Huang C-Y, Fang J-Y, Chen P-Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro Oncol. 2020;22(2):253–66.
Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, Güç E, Kapourani C-A, Byron A, Ferguson KM. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184(9):2454-2470.e2426.
Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33(2):312–9.
Zhang X, Chen L, Dang WQ, Cao MF, **ao JF, Lv SQ, Jiang WJ, Yao XH, Lu HM, Miao JY. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab Investig. 2020;100(4):619–29.
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18(3):519–27.
Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2(87):re6.
Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, De Groot JF. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012;14(11):1379–92.
Cui X, Morales RTT, Qian W, Wang H, Gagner JP, Dolgalev I, Placantonakis D, Zagzag D, Cimmino L, Snuderl M. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials. 2018;161:164–78.
Brandenburg S, Müller A, Turkowski K, Radev YT, Rot S, Schmidt C, Bungert AD, Acker G, Schorr A, Hippe A. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016;131(3):365–78.
Nijaguna MB, Patil V, Urbach S, Shwetha SD, Sravani K, Hegde AS, Chandramouli BA, Arivazhagan A, Marin P, Santosh V. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J Biol Chem. 2015;290(38):23401–15.
Urbantat RM, Jelgersma C, Brandenburg S, Nieminen-Kelhä M, Kremenetskaia I, Zollfrank J, Mueller S, Rubarth K, Koch A, Vajkoczy P. Tumor-associated microglia/macrophages as a predictor for survival in glioblastoma and temozolomide-induced changes in CXCR2 signaling with new resistance overcoming strategy by combination therapy. Int J Mol Sci. 2021;22(20):11180.
Li J, Kaneda MM, Ma J, Li M, Shepard RM, Patel K, Koga T, Sarver A, Furnari F, Xu B. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc Natl Acad Sci. 2021;118(16): e2009290118.
Chuang H-Y, Su Y-K, Liu H-W, Chen C-H, Chiu S-C, Cho D-Y, Lin S-Z, Chen Y-S, Lin C-M. Preclinical evidence of STAT3 inhibitor pacritinib overcoming temozolomide resistance via downregulating miR-21-enriched exosomes from M2 glioblastoma-associated macrophages. J Clin Med. 2019;8(7):959.
Li Z, Fu W-J, Chen X-Q, Wang S, Deng R-S, Tang X-P, Yang K-D, Niu Q, Zhou H, Li Q-R. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J Exp Clin Cancer Res. 2022;41(1):1–20.
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–86.
Aslan K, Turco V, Blobner J, Sonner JK, Liuzzi AR, Núñez NG, De Feo D, Kickingereder P, Fischer M, Green E. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat Commun. 2020;11(1):1–14.
Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, Vence L, Blando J, Zhao H, Yadav SS. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46.
Blank A, Kremenetskaia I, Urbantat RM, Acker G, Turkowski K, Radke J, Schneider UC, Vajkoczy P, Brandenburg S. Microglia/macrophages express alternative proangiogenic factors depending on granulocyte content in human glioblastoma. J Pathol. 2021;253(2):160–73.
Kim Y, Jeon H, Othmer H. The role of the tumor microenvironment in glioblastoma: a mathematical model. IEEE Trans Biomed Eng. 2016;64(3):519–27.
Su Y-T, Butler M, Zhang M, Zhang W, Song H, Hwang L, Tran AD, Bash RE, Schorzman AN, Pang Y. MerTK inhibition decreases immune suppressive glioblastoma-associated macrophages and neoangiogenesis in glioblastoma microenvironment. Neuro-oncol Adv. 2020;2(1):vdaa065.
Li Y, Guo S, Zhao K, Conrad C, Driescher C, Rothbart V, Schlomann U, Guerreiro H, Bopp MH, König A. ADAM8 affects glioblastoma progression by regulating osteopontin-mediated angiogenesis. Biol Chem. 2021;402(2):195–206.
De Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, Weathers S-P, Loghin M, Kamiya Matsouka C, Yung WA. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22(4):539–49.
Williamson LL, Chao A, Bilbo SD. Environmental enrichment alters glial antigen expression and neuroimmune function in the adult rat hippocampus. Brain Behav Immun. 2012;26(3):500–10.
Miyazaki T, Ishikawa E, Matsuda M, Sugii N, Kohzuki H, Akutsu H, Sakamoto N, Takano S, Matsumura A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kγ inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol. 2020;37(2):41–9.
Hsu SP, Chen Y-C, Chiang H-C, Huang Y-C, Huang C-C, Wang H-E, Wang Y-S, Chi K-H. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neurooncol. 2020;146(3):417–26.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253-267.e255.
Almahariq MF, Quinn TJ, Kesarwani P, Kant S, Miller CR, Chinnaiyan P. Inhibition of colony-stimulating factor-1 receptor enhances the efficacy of radiotherapy and reduces immune suppression in glioblastoma. In Vivo. 2021;35(1):119–29.
Zhu H, Leiss L, Yang N, Rygh CB, Mitra SS, Cheshier SH, Weissman IL, Huang B, Miletic H, Bjerkvig R. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget. 2017;8(7):12145.
Herting CJ, Chen Z, Maximov V, Duffy A, Szulzewsky F, Shayakhmetov DM, Hambardzumyan D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain. 2019;142(12):3834–51.
Pascual T, Villagrasa P, Vidal MJ, Ganau S, Bermejo B, Julve A, Zamora E, Miranda I, Vega E, Marquez C: Abstract OT1-01-01: SOLTI-1503 PROMETEO: Combination of talimogene laherparepvec (T-VEC) with atezolizumab in patients with residual breast cancer after standard neoadjuvant multi-agent chemotherapy. Cancer Research 2020, 80(4_Supplement):OT1-01-01-OT01-01-01.
Schuelke MR, Gundelach JH, Coffey M, West E, Scott K, Johnson DR, Samson A, Melcher A, Vile RG, Bram RJ. Phase I trial of sargramostim/pelareorep therapy in pediatric patients with recurrent or refractory high-grade brain tumors. Neuro-Oncol Adv. 2022;4(1):vdac85.
Zamarin D, Odunsi K, Slomovitz BM, Duska LR, Nemunaitis JJ, Reilley M, Bykov Y, Holland A, Hubbard-Lucey VM, Shohara L. Phase I/II study to evaluate systemic durvalumab+ intraperitoneal (IP) ONCOS-102 in patients with peritoneal disease who have epithelial ovarian (OC) or metastatic colorectal cancer (CRC): Interim phase I clinical and translational results. American Society of Clinical Oncology; 2020.
Zamarin D, Odunsi K, Zsiros E, Slomovitz BM, Pimentel A, Duska LR, Reilley M, Nemunaitis JJ, Hamouda DM, Patel H. Study to evaluate intraperitoneal (IP) ONCOS-102 with systemic durvalumab in patients with peritoneal disease who have epithelial ovarian (OC) or metastatic colorectal cancer (CRC): Phase 2 results. American Society of Clinical Oncology; 2022.
Wu J, Chen S, Baneyx G, Marchal A-L, Quinn DS, Martin M, Cervantes A, Sand-Dejmek J, Mataraza JM. Abstract LB061: On-target peripheral and tumor immune microenvironment modulation in patients treated with lacnotuzumab (anti-CSF1, MCS110)+ spartalizumab. Cancer Res. 2021;81(13_supplement):LB061.
Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. 2017;19(1):101–13.
Gomez-Roca CA, Cassier PA, Italiano A, Cannarile M, Ries C, Brillouet A, Mueller C, Jegg A-M, Meneses-Lorente G, Baehner M. Phase I study of RG7155, a novel anti-CSF1R antibody, in patients with advanced/metastatic solid tumors. American Society of Clinical Oncology; 2015.
Soto-Diaz K, Vailati-Riboni M, Louie AY, McKim DB, Gaskins HR, Johnson RW, Steelman AJ. Treatment with the CSF1R antagonist GW2580, sensitizes microglia to reactive oxygen species. Front Immunol. 2021;12:4889.
Neal ML, Fleming SM, Budge KM, Boyle AM, Kim C, Alam G, Beier EE, Wu LJ, Richardson JR. Pharmacological inhibition of CSF1R by GW2580 reduces microglial proliferation and is protective against neuroinflammation and dopaminergic neurodegeneration. FASEB J. 2020;34(1):1679–94.
Yan D, Kowal J, Akkari L, Schuhmacher A, Huse J, West B, Joyce J. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36(43):6049–58.
Crotty EE, Smith S, Brasel K, Pakiam F, Girard EJ, Connor YD, Zindy F, Mhyre AJ, Roussel MF, Olson JM. Medulloblastoma recurrence and metastatic spread are independent of colony-stimulating factor 1 receptor signaling and macrophage survival. J Neurooncol. 2021;153(2):225–37.
Zhu M, Bai L, Liu X, Peng S, **e Y, Bai H, Yu H, Wang X, Yuan P, Ma R. Silence of a dependence receptor CSF1R in colorectal cancer cells activates tumor-associated macrophages. J Immunother Cancer. 2022;10(12): e005610.
Piawah S, Hyland C, Umetsu SE, Esserman LJ, Rugo HS, Chien AJ. A case report of vanishing bile duct syndrome after exposure to pexidartinib (PLX3397) and paclitaxel. NPJ Breast Cancer. 2019;5(1):1–5.
Foster CC, Fleming GF, Karrison TG, Liao C-Y, Desai AV, Moroney JW, Ratain MJ, Nanda R, Polite BN, Hahn OM. Phase I study of stereotactic body radiotherapy plus nivolumab and urelumab or cabiralizumab in advanced solid tumors SBRT and dual immunotherapy for advanced solid tumors. Clin Cancer Res. 2021;27(20):5510–8.
Irenaeus SM, Nielsen D, Ellmark P, Yachnin J, Deronic A, Nilsson A, Norlén P, Veitonmäki N, Wennersten CS, Ullenhag GJ. First-in-human study with intratumoral administration of a CD40 agonistic antibody, ADC-1013, in advanced solid malignancies. Int J Cancer. 2019;145(5):1189–99.
Barlesi F, Lolkema M, Rohrberg KS, Hierro C, Marabelle A, Razak AA, Teixeira L, Boni V, Miller WH, Aggarwal C. 291 Phase Ib study of selicrelumab (CD40 agonist) in combination with atezolizumab (anti-PD-L1) in patients with advanced solid tumors. BMJ Spec J. 2020. https://doi.org/10.1136/jitc-2020-SITC2020.0291.
Machiels J-P, Gomez-Roca C, Michot J-M, Zamarin D, Mitchell T, Catala G, Eberst L, Jacob W, Jegg A-M, Cannarile MA. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J Immunother Cancer. 2020;8(2):e001153.
Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, Kerr P, Chan C, Smith A, Steven N. Clinical and biological effects of an agonist anti-CD40 antibody: a cancer research Uk phase I study anti-CD40 phase I study. Clin Cancer Res. 2015;21(6):1321–8.
Grilley-Olson JE, Curti BD, Smith DC, Goel S, Gajewski T, Markovic S, Rixe O, Bajor DL, Gutierrez M, Kuzel T. SEA-CD40, a non-fucosylated CD40 agonist: Interim results from a phase 1 study in advanced solid tumors. American Society of Clinical Oncology; 2018.
Lindsay H, Onar-Thomas A, Kocak M, Poussaint TY, Dhall G, Broniscer A, Vinitsky A, MacDonald T, Trifan O, Fangusaro J. EPCT-02. PBTC-051: first in pediatrics phase 1 study of CD40 agonistic monoclonal antibody APX005M in pediatric subjects with recurrent/refractory brain tumors. Neuro Oncol. 2020;22(Suppl 3):304.
Desjardins A, Chandramohan V, Landi DB, Johnson MO, Khasraw M, Peters KB, Low J, Herndon JE, Threatt S, Bullock CA. A phase 1 trial of D2C7-it in combination with an Fc-engineered anti-CD40 monoclonal antibody (2141–V11) administered intratumorally via convection-enhanced delivery for adult patients with recurrent malignant glioma (MG). American Society of Clinical Oncology; 2022.
Sanford N, Elghonaimy E, Kardosh A, Kazmi S, Pogacnik JS, Yang X, Timmerman R, Aguilera T. 411 INNATE: immunotherapy during neoadjuvant therapy for rectal cancer to elucidate local and systemic therapeutic responses. BMJ Spec J. 2021.
Zakharia Y, Rixe O, Ward JH, Drabick JJ, Shaheen MF, Milhem MM, Munn D, Kennedy EP, Vahanian NN, Link CJ. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. American Society of Clinical Oncology; 2018.
Zakharia Y, McWilliams RR, Rixe O, Drabick J, Shaheen MF, Grossmann KF, Kolhe R, Pacholczyk R, Sadek R, Tennant LL. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J ImmunoTher Cancer. 2021;9(6):e002057.
Pu J, Xu Z, Nian J, Fang Q, Yang M, Huang Y, Li W, Ge B, Wang J, Wei H. M2 macrophage-derived extracellular vesicles facilitate CD8+ T cell exhaustion in hepatocellular carcinoma via the miR-21–5p/YOD1/YAP/β-catenin pathway, vol. 7. Nature Publishing Group; 2021. p. 1–14.
Kotecki N, Vuagnat P, O’Neil BH, Jalal S, Rottey S, Prenen H, Benhadji KA, **a M, Szpurka AM, Saha A. A phase I study of an IDO-1 inhibitor (LY3381916) as monotherapy and in combination with an anti-PD-L1 antibody (LY3300054) in patients with advanced cancer. J Immunother. 2021;44(7):264–75.
Gao J, Deng F, Jia W. Inhibition of indoleamine 2, 3-dioxygenase enhances the therapeutic efficacy of immunogenic chemotherapeutics in breast cancer. J Breast Cancer. 2019;22(2):196–209.
Jung KH, LoRusso P, Burris H, Gordon M, Bang Y-J, Hellmann MD, Cervantes A, Ochoa de Olza M, Marabelle A, Hodi FS. Phase I study of the indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (atezolizumab) in advanced solid tumors navoximod and atezolizumab in advanced solid tumors. Clin Cancer Res. 2019;25(11):3220–8.
Yap TA, Sahebjam S, Hong DS, Chiu VK, Yilmaz E, Efuni S, Grebennik DO, Collaku A, Ogunmefun E, Liu Y. First-in-human study of KHK2455, a long-acting, potent and selective indoleamine 2, 3-dioxygenase 1 (IDO-1) inhibitor, in combination with mogamulizumab (Moga), an anti-CCR4 monoclonal antibody, in patients (pts) with advanced solid tumors. American Society of Clinical Oncology; 2018.
Johnson T, Aguilera D, Al-Basheer A, Berrong Z, Castellino R, Eaton B, Esiashvili N, Foreman N, Heger I, Kennedy E. Results of the NLG2105 phase I trial using the IDO pathway inhibitor indoximod, in combination with radiation and chemotherapy, for children with newly diagnosed DIPG. Ann Oncol. 2019;30:xi38.
Reardon DA, Desjardins A, Rixe O, Cloughesy T, Alekar S, Williams JH, Li R, Taylor CT, Lassman AB. A phase 1 study of PF-06840003, an oral indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor in patients with recurrent malignant glioma. Invest New Drugs. 2020;38(6):1784–95.
Chen B, Alvarado DM, Iticovici M, Kau NS, Park H, Parikh PJ, Thotala D, Ciorba MA. Interferon-induced IDO1 mediates radiation resistance and is a therapeutic target in colorectal cancer IDO1 inhibition synergizes with radiotherapy. Cancer Immunol Res. 2020;8(4):451–64.
Shi J, Liu C, Luo S, Cao T, Lin B, Zhou M, Zhang X, Wang S, Zheng T, Li X. STING agonist and IDO inhibitor combination therapy inhibits tumor progression in murine models of colorectal cancer. Cell Immunol. 2021;366: 104384.
Shen F, Feng L, Zhu Y, Tao D, Xu J, Peng R, Liu Z. Oxaliplatin-/NLG919 prodrugs-constructed liposomes for effective chemo-immunotherapy of colorectal cancer. Biomaterials. 2020;255: 120190.
Papadopoulos KP, Tsai FY-C, Bauer TM, Muigai L, Liang Y, Bennett MK, Orford KW, Fu S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. American Society of Clinical Oncology; 2017
Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5(1):1–18.
Avtandilyan N, Javrushyan H, Mamikonyan A, Grigoryan A, Trchounian A. The potential therapeutic effect of NG-hydroxy-nor-L-arginine in 7, 12-dimethylbenz (a) anthracene-induced breast cancer in rats. Exp Mol Pathol. 2019;111: 104316.
Lucas J, Hsieh T-C, Halicka HD, Darzynkiewicz Z, Wu JM. Upregulation of PD-L1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300-mediated NF-κB signaling. Int J Oncol. 2018;53(4):1469–80.
Zeng A, Liang X, Zhu S, Liu C, Wang S, Zhang Q, Zhao J, Song L. Chlorogenic acid induces apoptosis, inhibits metastasis and improves antitumor immunity in breast cancer via the NF-κB signaling pathway. Oncol Rep. 2021;45(2):717–27.
Azambuja JH, Ludwig N, Yerneni SS, Braganhol E, Whiteside TL. Arginase-1+ exosomes from reprogrammed macrophages promote glioblastoma progression. Int J Mol Sci. 2020;21(11):3990.
Pilanc P, Wojnicki K, Roura A-J, Cyranowski S, Ellert-Miklaszewska A, Ochocka N, Gielniewski B, Grzybowski MM, Błaszczyk R, Stańczak PS. A novel oral arginase 1/2 inhibitor enhances the antitumor effect of PD-1 inhibition in murine experimental gliomas by altering the immunosuppressive environment. Front Oncol. 2021;11:703465.
Hou N, Liu N, Han J, Yan Y, Li J. Chlorogenic acid induces reactive oxygen species generation and inhibits the viability of human colon cancer cells. Anticancer Drugs. 2017;28(1):59–65.
Tawbi HAH, Peng W, Phillips S, Milton DR, Amaria RN, Diab A, Glitza IC, Patel SP, Wong MK, Yee C. Safety results from phase I/II study of the PI3Kβ inhibitor GSK2636771 (G) in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. American Society of Clinical Oncology; 2020.
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH. A Phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− Metastatic breast cancer alpelisib and letrozole in ER+ metastatic breast cancer. Clin Cancer Res. 2017;23(1):26–34.
Narayan P, Prowell TM, Gao JJ, Fernandes LL, Li E, Jiang X, Qiu J, Fan J, Song P, Yu J. FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin Cancer Res. 2021;27(7):1842–9.
Juric D, Kalinsky K, Oliveira M, Cervantes A, Bedard P, Krop I, Hamilton E, Schmid P, Varga A, Turner N. Abstract OT1-08-04: A first-in-human phase Ia dose escalation study of GDC-0077, a p110a-selective and mutant-degrading PI3K inhibitor, in patients with PIK3CA-mutant solid tumors. Cancer Res. 2020; 80(4_Supplement):OT1-08-04-OT01-08-04.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA. The identification of 2-(1 H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno [3, 2-d] pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–32.
Harder BG, Peng S, Sereduk CP, Sodoma AM, Kitange GJ, Loftus JC, Sarkaria JN, Tran NL. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol Med. 2019;25(1):1–11.
Zhang X, Li W, Sun J, Yang Z, Guan Q, Wang R, Li X, Li Y, Feng Y, Wang Y. How to use macrophages to realise the treatment of tumour. J Drug Target. 2020;28(10):1034–45.
Hainsworth JD, Becker KP, Mekhail T, Chowdhary SA, Eakle JF, Wright D, Langdon RM, Yost KJ, Padula GDA, West-Osterfield K. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J Neurooncol. 2019;144(2):303–11.
Bowles DW, Kochenderfer M, Cohn A, Sideris L, Nguyen N, Cline-Burkhardt V, Schnadig I, Choi M, Nabell L, Chaudhry A. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin Colorectal cancer. 2016;15(4):337-344.e332.
Bardia A, Gounder M, Rodon J, Janku F, Lolkema MP, Stephenson JJ, Bedard PL, Schuler M, Sessa C, LoRusso P. Phase Ib study of combination therapy with mek inhibitor binimetinib and phosphatidylinositol 3-kinase inhibitor buparlisib in patients with advanced solid tumors with RAS/RAF alterations. Oncologist. 2020;25(1):e160–9.
Landry M, DuRoss A, Neufeld M, Hahn L, Sahay G, Luxenhofer R, Sun C. Low dose novel PARP-PI3K inhibition via nanoformulation improves colorectal cancer immunoradiotherapy. Mater Today Bio. 2020;8: 100082.
Hu Y, Zhang K, Zhu X, Zheng X, Wang C, Niu X, Jiang T, Ji X, Zhao W, Pang L. Synergistic inhibition of drug-resistant colon cancer growth with PI3K/mTOR dual inhibitor BEZ235 and nano-emulsioned paclitaxel via reducing multidrug resistance and promoting apoptosis. Int J Nanomed. 2021;16:2173.
Alqurashi N, Hashimi SM, Alowaidi F, Ivanovski S, Wei MQ. Dual mTOR/PI3K inhibitor NVP-BEZ235 arrests colorectal cancer cell growth and displays differential inhibition of 4E-BP1. Oncol Rep. 2018;40(2):1083–92.
Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34.
Lipson E, Long G, Tawbi H, Schadendorf D, Atkinson V, Maurer M, Simonsen K, Harbison C, Hodi F. CA224–047: a randomized, double-blind, phase II/III study of relatlimab (anti–LAG-3) in combination with nivolumab (anti–PD-1) versus nivolumab alone in previously untreated metastatic or unresectable melanoma. Ann Oncol. 2018;29:viii464–5.
Dirix L, Triebel F. AIPAC: a phase IIb study of eftilagimod alpha (IMP321 or LAG-3Ig) added to weekly paclitaxel in patients with metastatic breast cancer. Future Oncol. 2019;15(17):1963–73.
Wildiers H, Armstrong A, Cuypere E, Dalenc F, Dirix L, Chan S, Marme F, Schröder CP, Huober J, Wagemans J. Abstract PD14–08: Primary efficacy results from AIPAC: A double-blinded, placebo controlled, randomized multinational phase IIb trial comparing weekly paclitaxel plus eftilagimod alpha (soluble LAG-3 protein) vs. weekly paclitaxel plus placebo in HR-positive metastatic breast cancer patients. Cancer Res 2021, 81(4_Supplement):PD14-08-PD14-08.
Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, Chien AJ, Forero-Torres A, Ellis E, Han H. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol. 2020;6(5):676–84.
Harris-Bookman S, Mathios D, Martin AM, **a Y, Kim E, Xu H, Belcaid Z, Polanczyk M, Barberi T, Theodros D. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–8.
Mair M, Kiesel B, Feldmann K, Widhalm G, Dieckmann K, Woehrer A, Muellauer L, Preusser M, Berghoff AS. Lymphocyte-activation gene 3 (LAG-3) expression in the inflammatory microenvironment of glioma. American Society of Clinical Oncology; 2020.
Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im S-A, Golan T, Geva R, Wermke M, De Miguel M. A phase 1 first-in-human study of the anti-LAG-3 antibody MK4280 (favezelimab) plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. J Clin Oncol. 2021;39:3584.
Garralda E, Sukari A, Lakhani N, Patnaik A, Lou Y, Im S-A, Golan T, Geva R, Wermke M, de Miguel M. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open. 2022;7(6): 100639.
Moore PA, Shah K, Yang Y, Alderson R, Roberts P, Long V, Liu D, Li JC, Burke S, Ciccarone V. Development of MGD007, a gpA33 x CD3-bispecific DART protein for T-cell immunotherapy of metastatic colorectal cancer. Mol Cancer Ther. 2018;17(8):1761–72.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
The core idea of this study came from ZP, MNA and BM. ZP, MNA and BM directed the other authors and analyzed the collected papers. All authors wrote the manuscript. Final editing was done by ZP, RKP, and MNA. MNA and ZP supervised the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
There is no involvement of human or animal in this study.
Consent for publication
Not applicable.
Competing interests
There are neither ethical nor financial conflicts of interest involved in the manuscript. The manuscript is not submitted for publication elsewhere.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jahandideh, A., Yarizadeh, M., Noei-Khesht Masjedi, M. et al. Macrophage’s role in solid tumors: two edges of a sword. Cancer Cell Int 23, 150 (2023). https://doi.org/10.1186/s12935-023-02999-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02999-3