
Abstract
Background
Both N6-methyladenosine (m6A) modification and lncRNAs play an important role in the carcinogenesis and cancer inhibition of ovarian cancer (OC). However, lncRNAs involved in m6A regulation (LI-m6As) have never been reported in OC. Herein, we aimed to identify and validate a signature based on LI-m6A for OC.
Methods
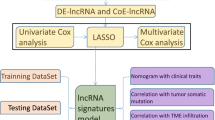
RNA sequencing profiles with corresponding clinical information associated with OC and 23 m6A regulators were extracted from TCGA. The Pearson correlation coefficient (PCC) between lncRNAs and 23 m6A regulators (|PCC|> 0.4 and p < 0.01) was calculated to identify LI-m6As. The LI-m6As with significant prognostic value were screened based on univariate Cox regression analysis to construct a risk model by LASSO Cox regression. Gene Set Enrichment Analysis (GSEA) was implemented to survey the biological functions of the risk groups. Several clinicopathological characteristics were utilized to evaluate their ability to predict prognosis, and a nomogram was constructed to evaluate the accuracy of survival prediction. Besides, immune microenvironment, checkpoint, and drug sensitivity in the two risk groups were compared using comprehensive algorithms. Finally, real-time qPCR analysis and cell counting kit-8 assays were performed on an alternative lncRNA, CACNA1G-AS1.
Results
The training cohort involving 258 OC patients and the validation cohort involving 111 OC patients were downloaded from TCGA. According to the PCC between the m6A regulators and lncRNAs, 129 LI-m6As were obtained to perform univariate Cox regression analysis and then 10 significant prognostic LI-m6As were identified. A prognostic signature containing four LI-m6As (AC010894.3, ACAP2-IT1, CACNA1G-AS1, and UBA6-AS1) was constructed according to the LASSO Cox regression analysis of the 10 LI-m6As. The prognostic signature was validated to show completely opposite prognostic value in the two risk groups and adverse overall survival (OS) in several clinicopathological characteristics. GSEA indicated that differentially expressed genes in disparate risk groups were enriched in several tumor-related pathways. At the same time, we found significant differences in some immune cells and chemotherapeutic agents between the two groups. An alternative lncRNA, CACNA1G-AS1, was proven to be upregulated in 30 OC specimens and 3 OC cell lines relative to control. Furthermore, knockdown of CACNA1G‐AS1 was proven to restrain the multiplication capacity of OC cells.
Conclusions
Based on the four LI-m6As (AC010894.3, ACAP2-IT1, CACNA1G-AS1, and UBA6-AS1), the risk model we identified can independently predict the OS and therapeutic value of OC. CACNA1G‐AS1 was preliminarily proved to be a malignant lncRNA.
Similar content being viewed by others
Background
Ovarian cancer (OC) is the main cause of gynecological cancer-related death worldwide [1]. Because of the lack of clinical manifestations in the early stage, 70% of patients are diagnosed in the middle or late stages [1]. It is also difficult to perform radical surgery, leading to a high OC mortality rate of OC [1]. OC is characterized by obvious tissue heterogeneity with genomic characteristics, which shows that it easily develops resistance to chemotherapy and leads to a high tumor recurrence rate [1]. Therefore, improving the prognosis of OC patients by exploring novel diagnoses and therapies is an urgent problem to address.
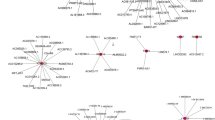
Studies have shown that genome-level epigenetic modifications, such as DNA methylation, histone modification and RNA editing, are crucial to tumorigenesis. N6-methyladenosine (m6A) modification was first detected in the poly (A) RNA component in 1974 and is considered to be the most abundant posttranscriptional modification of mRNA [2]. m6A modification is mainly associated with three types of proteases that serve as regulators, namely, methyltransferases (writers), demethylases (erasers), and signal transducers (readers) [2]. Numerous studies have shown that different levels of m6A regulators are associated with the self-renewal of tumor stem cells, the proliferation of cancer cells, and the sensitivity to chemotherapy [2]. A previous study showed that high-frequency genetic alterations of m6A RNA methylation regulators are crucial for the progression [3] and that m6A modification contributes to PARPi resistance in OC [4]. In OC, YTHDF1 (reader) facilitates the expression of EIF3C [5] and the stem cell-like phenotype of cisplatin resistance [6] in a m6A-dependent manner, thereby strengthening tumorigenesis and metastasis. ALKBH5 (eraser) [7] and YTHDF2 (reader) [8, 9] were shown to regulate the carcinogenesis of OC by modulating m6A levels. IGF2BP1 (reader) was demonstrated to augment the translation of serum response factor through m6A modification [10]. METTL3 (writer) can regulate m6A methylation and thus regulate the malignancy of OC [11,3a, Additional file 1: Table S1). Interestingly, p53 may regulate m6A factors and LI-m6As through trans interactions (p53-IGF2BP1-AC010745.4/CACNA1G-AS1/ERVH48-1/HOXA-AS3/PTENP1-AS). On the basis of ten lncRNA-m6A pairs, we further found four of them included in the lncRNA-miRNA-mRNA relationship pairs and the top one lncRNA-m6A pair with most miRNAs was UBA6-AS1-YTHDC1(Fig. 3b). These findings may provide insights into the regulatory mechanisms of lncRNAs correlated with m6A-regulatory genes.

Network based on trans interactions and ceRNA. a Trans interaction for lncRNA-TF-mRNA relationship pairs. b ceRNA for lncRNA-miRNA-mRNA relationship pairs. Blue arrowheads: TFs; red rhombus: LI-m6As; yellow ellipses: m6A regulators; blue rectangles: miRNAs
Construction and validation the prognostic signature
A LI-m6A prognostic signature was constructed according to the LASSO Cox analysis of 10 significant prognostic LI-m6As. The λ selection diagram is shown in Fig. 4a, b. λ between λmin and λ1se were considered appropriate. The model constructed by λ1se was the simplest, that was, it used a small number of genes, while λmin had a higher accuracy rate and used a larger number of genes. The λmin was selected to build the model for accuracy in our study. Patients in the training and validation cohorts were divided into low- or high-risk subgroups based on the median of RSs. The K–M survival curves of risk groups revealed that OS in the high-risk group was markedly lower than that in the low-risk group in both cohorts (Fig. 5a, b). The ROC curves proved that the risk model signature based on four m6A-RLs was accurate in predicting the OS of OC patients (Fig. 5c, d).

a LASSO Cox analysis of 10 significant prognostic LI-m6As. b λ selection diagram. The two dotted lines indicated two particular values of λ. The left side was λmin and the right side was λ1se. The λmin was selected to build the model for accuracy in our study

Construction and validation of the prognostic signature. a, b The K-M survival curves of two risk groups based on the risk model in the training cohort (a) and validation cohort (d). c, d The ROC curves for predicting 1-, 3-, and 5-year survival in in the training cohort (c) and validation cohort (d)
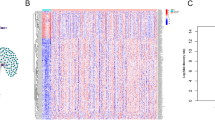
Four LI-m6As with corresponding coefficients and HR values (Table 2), namely, AC010894.3, ACAP2-IT1, CACNA1G-AS1, and UBA6-AS1, were generated to calculate RSs.. A heatmap of the associations between the expression levels of the four LI-m6As and RSs showed that the expression of ACAP2-IT1 and CACNA1G-AS1 increased with increasing RS, whereas the expression of AC010894.3 and UBA6-AS1 decreased with increasing RS in both the training and validation cohorts (Fig. 6a, b). The K–M survival curves confirmed that lower expression of ACAP2-IT1 and CACNA1G-AS1 and higher expression of AC010894.3 and UBA6-AS1 were associated with better OS of OC patients (Fig. 6c-f). Hence, ACAP2-IT1 and CACNA1G-AS1 were risk factors, while AC010894.3 and UBA6-AS1 were protective factors.

Prognostic analysis of the four LI-m6As. a, b Heatmap of the expression levels of the four m6A-RLs with RSs in the training (a) and validation cohort (b). c-f The K-M survival curves of LI-m6A AC010894.3 (c), ACAP2-IT1 (d), CACNA1G-AS1 (e), and UBA6-AS1 (f)
Nomogram model construction and visualization
Univariate and multivariate Cox regression analyses of clinicopathology and the LI-m6A risk model showed that the signature was an independent risk factor for OC patients in the training and validation cohorts (Table 3). A nomogram was further constructed based on risk score, age, grade, and Figo state to predict the prognosis of OC (Fig. 7a, b). A calibration diagram was also used to verify the prediction ability of risk score for 1-, 3- and 5-year of survival (Fig. 7c–h). In the calibration plots, the blue solid line was the prediction for survival, and the diagonal dotted line was the actual survival. The closer the solid line was to the dotted line, the better the prediction ability was.

Nomogram model construction and visualization. a, b The Nomogram model based on risk score and clinical features for the training (a) and validation (b) cohort. c-h The calibration plots of the nomogram for predicting the probability of OS at 1 (c, f), 3 (d, g), and 5 (e, h) years in the training cohort (c–e) and validation cohort (f–h)
Clinicopathology analysis
We carried out clinicopathological analysis to evaluate the prognostic capacity of the LI-m6As prognostic signature. Several clinicopathological characteristics, such as WHO grade I, grade IV and FIGO stage I, were excluded due to the small sample size (< 10 patients). The results showed that the OS of the clinicopathology subgroups, such as Grade II, Grade III, age < = 55 or > 55 years old, Stage III, Stage IV, platinum sensitivity and platinum resistance, were significantly different in the low- or high-risk group (Fig. 8a-h). Potentially, the p value of the Stage II subgroup was indistinctive because of insufficient sample size (Additional file 2: Fig. S1).

The K-M survival curves of several clinicopathological characteristics based on the risk model. a Grade II. b Grade III. c Age < = 55. d Age > 55. e Stage III. f Stage IV. g Platinum sensitivity. h Platinum resistance
Biofunctional analysis of the risk groups
A total of 1581 DEGs between the risk subgroups were identified. The GO biological process analysis found that DEGs were mainly enriched in migration and proliferation, such as epithelial cell migration and vascular smooth muscle cell proliferation (Fig. 9a). Moreover, the performed KEGG pathway analysis revealed that the DEGs were mainly enriched in microRNAs in cancer and taste transduction (Fig. 9b). GSEA of GO indicated that gene sets enriched in ‘negative regulation of gene expression’, ‘negative regulation of metabolic process’, and ‘posttranscriptional regulation of gene expression’ were highly expressed in the high-risk group (Fig. 9c). GSEA of KEGG showed that genes enriched in ‘MicroRNAs in cancer’ were highly expressed the in high-risk group, while genes enriched in ‘Metabolic pathways’ were expressed at low levels in the high-risk group (Fig. 9d). The enriched GO terms and KEGG pathways were further annotated from “Metascape” website (Fig. 9e). The key module of the PPI network discerned by MCODE were shown in Fig. 9f. Seven hub genes were included in the interaction network.

Functional analysis. a, b Significantly enriched GO terms (a) and KEGG pathways (b) of differentially expressed genes (DEGs). The color scale represented p value and the circle size indicated count. c, d Gene set enrichment analysis (GSEA) of GO (c) and KEGG (d). The crest on the left represents the high expression of the enriched genes in the high-risk group, while the crest on the right is the opposite. e Cluster ID. Nodes with the same cluster ID are typically close to each other and the same color indicates the same cluster ID. f The key module of PPI network. Seven hub genes were included in the interaction network. Circles indicate the genes in the PPI network, and the connection indicates the potential interaction between different mRNAs
Immune microenvironment and checkpoint
According to five algorithms (Estimate, ssGSEA, Cibersort, MCPcounter and xCell), Stromal Score, Immune Score, ESTIMATE Score, and relative infiltration abundance of immune cells and stromal cells of each sample were estimated respectively. The results revealed that a total of 59 microenvironment cells and Stromal score had significant differences between the two groups (Fig. 10a, Additional file 3: Table. S2). Among them, Neutrophils cells, Fibroblasts cells, and Endothelial cells were the top 3 immune cells significantly enriched in the high-risk group. Given the importance of immune checkpoints in cancer treatment, the expressions of seven checkpoint genes were compared. We found that HAVCR2 and SIRPA had higher expressions in the low-risk group (Fig. 10b). In Fig. 10c, OC patients in low-risk group exhibited higher TIDE scores than those in high-risk group, indicating that OC patients with lower RSs were more sensitive to ICB therapy.

Immune microenvironment and checkpoint. a Heatmap of immune microenvironment revealed that a total of 59 immune cells and stromal score had significant differences between the two risk groups. b Expression of seven immune checkpoint genes between high and low-risk group. HAVCR2 and SIRPA had higher expressions in the low-risk group. c TIDE scores in the low-risk group were higher than those in the high-risk group. Data are shown as means ± S.D. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Sensitivity of chemotherapy drug
The IC50 levels of 135 drugs in OC patients were quantified and significant differences in 87 chemotherapy drugs were found between the risk groups (Additional file 4: Table. S3). Figure 11 exhibited the results of nine commonly used chemotherapeutic agents for OC. Our data showed that the IC50 levels of Cisplatin (Fig. 11a) were significantly higher in high-risk group than that in low-risk group. Inversely, the IC50 levels of Bleomycin (Fig. 11b), Docetaxel (Fig. 11c), Gemcitabine (Fig. 11d), Vinblastine (Fig. 11e) and Vinorelbine (Fig. 11f) were significantly lower in high-risk group than that in low-risk group, indicating that the OC patients in the high-risk group were more sensitive to these drugs. However, the sensitivity of the two risk groups to Paclitaxel (Fig. 11g), Rucaparib (Fig. 11h) and Veliparib (Fig. 11i) did not show a significant difference.

Sensitivity of chemotherapy drug. a-i Difference in the estimated IC50 levels of Cisplatin (a), Bleomycin (b), Docetaxel (c), Gemcitabine (d), Vinblastine (e), Vinorelbine (f), Paclitaxel (g), Rucaparib (h) and Veliparib (i). Data are shown as means ± S.D. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
In vitro assays
lncRNA CACNA1G-AS1 was filtered as the candidate molecule to perform cell function assays. Real-time qPCR analysis indicated that CACNA1G-AS1 was significantly upregulated in 30 OC tissues and three OC cell lines (Fig. 12a, b). The transfection efficiencies of three siRNAs targeting CACNA1G-AS1 were detected, which revealed that siRNA-2 presented the highest transfection efficiency in SKOV-3 cells (Fig. 12c) and that siRNA-3 presented the highest transfection efficiency in A2780 cells (Fig. 12e). The CCK-8 assay showed that the viability of SKOV-3 (Fig. 12d) and A2780 cells (Fig. 12f) was suppressed after transfection of the corresponding siRNAs.

Cell function assays. a, b Real-time qPCR analysis detecting relative expression of lncRNA CACNA1G-AS1 in 30 OC tissues (a) and three OC cell lines (b). c The transfection efficacities of three siRNAs targeting CACNA1G-AS1 in SKOV-3 cells. d CCK-8 assays were performed after CACNA1G-AS1 was inhibited in SKOV-3. e The transfection efficacities of three siRNAs targeting CACNA1G-AS1 in A2780. f CCK-8 assays were performed after CACNA1G-AS1 was inhibited in A2780. Data are shown as means ± S.D. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Discussion
As the most common internal modification of mRNAs in all higher eukaryotes, m6A has been proven to be abnormally expressed in a variety of tumors, and plays an important role in the regulation of a series of malignant biological behaviors such as cell proliferation, invasion and metastasis [2]. Previous studies have reported that several lncRNAs regulate the occurrence and development of cancer via m6A modification. For example, lncRNA RP11-138 J23.1 regulated by m6A positively induced the epithelial mesenchymal transition (EMT) of colorectal cancer cells [38]. Oncogenic lncRNA XIST is inhibited by METTL14 (writer) in colorectal cancer [39]. The lncRNA DANCR and IGF2BP2 (reader) have been proven to synergistically promote the pathogenesis of pancreatic cancer [40]. It has been unveiled that LINC00942 serves as an oncogene by promoting METTL14-mediated m6A methylation in breast cancer [41]. lncRNA GAS5 negatively regulated by YTHDF3 (reader) was demonstrated to be a tumor-suppressor in colorectal cancer [42]. LINRIS was proven to promote carcinogenesis of colorectal cancer by serving as a stabilizer of IGF2BP2 (reader) [43]. LNCAROD is aberrantly expressed and has been identified as an oncogenic lncRNA due to dysregulation of m6A modification [44]. The abovementioned evidence shows that m6A is modified in the process of lncRNA biological function. In addition, lncRNA can also regulate m6A modification. These are all part of the complex regulatory network of tumors. As a new discovery in scientific research, we believe that in-depth study of the biological mechanism of mutual regulation between lncRNAs and m6A will become a hot spot for discovering prognostic markers or therapeutic targets for malignant tumors. Nonetheless, lncRNAs involved in m6A regulation in OC are still unknown.
Four LI-m6As, namely, AC010894.3, ACAP2-IT1, CACNA1G-AS1, and UBA6-AS1, were included in our identified prognostic signature. They were correlated with METTL5 (writer), RBM15 (writer), IGF2BP1 (reader), and YTHDC1 (reader), respectively. Among the four m6A regulators, METTL5 was confirmed to serve as the enzyme for the m6A modification of 18S rRNA, thereby promoting breast cancer cell growth [45]. RBM15 was identified as a regulator that binds to METTL3 and WTAP and directs these two proteins to specific RNA sites for m6A modification [2]. Studies have shown that lncRNAs participate in mediating the occurrence and progression of cancer by targeting IGFBP1, thus becoming a new therapeutic target for cancer [46]. The m6A level of lncRNA pncRNA-D was recognized by YTHDC1 [47]. For the LI-m6As we identified, a study has shown that UBA6-AS1 serves as a malignant gene in glioblastoma by competitively binding to miR-7648. Previous studies have demonstrated that lncRNA CACNA1G-AS1 enhances the multiplication capacity via the miR-2392/C1orf61 pathway in hepatocellular cancer [49]. CACNA1G-AS1 promoted the proliferation and invasiveness of colorectal cancer by inhibiting the expression of p53 [50]. CACNA1G-AS1 exerts its malignant function by HNRNPA2B1 in non-small-cell lung cancer [51]. In our research, CACNA1G-AS1 showed high expression and may regulate proliferation, which would fill the gap in the OC literature. Our findings are probably worthy of further study by subsequent researchers who concentrate on the mechanism of m6A-related lncRNAs in OC.
We further explored the differences in immune microenvironment, immune checkpoint, chemotherapy drug sensitivity between the two risk groups. Our data showed that OC patients in the low-risk group may be more sensitive to Cisplatin and ICB therapy compared to the high-risk group, while patients in the high-risk group were more sensitive to Bleomycin, Docetaxel, Gemcitabine, Vinblastine and Vinorelbine. Findings of our study uncovered potential biomarker and therapeutic target for the risk model based on the LI-m6As.
Our study still has some limitations. First, there were only 30 OC patients without OS in our cohort, hence, more time and more samples are needed for follow-up. Second, cell function assays of CACNA1G-AS1 were preliminary and need further investigation to provide a better understanding. Definitively, the number of OC samples in TCGA is very limited; hence, more independent data sets are needed to validate our identified LI-m6As.
Conclusions
We found four lncRNAs (AC010894.3, ACAP2-IT1, CACNA1G-AS1, and UBA6-AS1) involved in m6A regulation and that have significant prognostic value in OC. Furthermore, the predictive signature based on the four lncRNAs can independently predict the therapeutic value of OC patients by combining molecular signatures and clinical characteristics. Furthermore, CACNA1G-AS1 was preliminarily identified as a malignant lncRNA through in vitro experiments.
Availability of data and materials
The RNA sequencing profiles are able to be gained from The Cancer Genome Atlas (TCGA) (https://toil.xenahubs.net). Further inquiries can be directed to the corresponding author.
Abbreviations
- DEG:
-
Differentially expressed genes
- GO:
-
Gene ontology
- GRCh38:
-
Genome Reference Consortium Human Build 38
- GSEA:
-
Gene Set Enrichment Analysis
- HR:
-
Hazard ratio
- K-M:
-
Kaplan–Meier
- LASSO:
-
Least absolute shrinkage and selection operator
- LI-m6As:
-
LncRNA involved in m6A regulation
- lncRNA:
-
Long noncoding RNA
- m6A:
-
N6-methyladenosine
- OC:
-
Ovarian cancer
- OS:
-
Overall survival
- PPI:
-
Protein–protein interaction
- RS:
-
Risk score
- TCGA:
-
The Cancer Genome Atlas
- TIDE:
-
Tumor Immune Dysfunction and Exclusion
References
Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. J Clin. 2019. 69(4):280–304.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88.
Han X, Liu J, Cheng G, Cui S. Gene Signatures and Prognostic Values of m6A RNA Methylation Regulators in Ovarian Cancer. Cancer Control J Moffitt Cancer Center. 2020;27(1):1073274820960460.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, Liu P, Kossenkov AV, Showe LC, Jean S, et al. N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Can Res. 2019;79(11):2812–20.
Liu T, Wei Q, ** J, Luo Q, Liu Y, Yang Y, Cheng C, Li L, Pi J, Si Y, et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48(7):3816–31.
Hao L, Wang JM, Liu BQ, Yan J, Li C, Jiang JY, Zhao FY, Qiao HY, Wang HQ. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochimica et biophysica acta Mol Cell Res. 2021;1868(1):118878.
Jiang Y, Wan Y, Gong M, Zhou S, Qiu J, Cheng W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-κB pathway. J Cell Mol Med. 2020;24(11):6137–48.
Li J, Wu L, Pei M, Zhang Y. YTHDF2, a protein repressed by miR-145, regulates proliferation, apoptosis, and migration in ovarian cancer cells. J Ovarian Res. 2020;13(1):111.
Xu F, Li J, Ni M, Cheng J, Zhao H, Wang S, Zhou X, Wu X. FBW7 suppresses ovarian cancer development by targeting the N(6)-methyladenosine binding protein YTHDF2. Mol Cancer. 2021;20(1):45.
Müller S, Glaß M, Singh AK, Haase J, Bley N, Fuchs T, Lederer M, Dahl A, Huang H, Chen J, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47(1):375–90.
Ma Z, Li Q, Liu P, Dong W, Zuo Y. METTL3 regulates m6A in endometrioid epithelial ovarian cancer independently of METTl14 and WTAP. Cell Biol Int. 2020;44(12):2524–31.
Hua W, Zhao Y, ** X, Yu D, He J, **e D, Duan P. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–65.
Liang S, Guan H, Lin X, Li N, Geng F, Li J. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol Lett. 2020;19(4):3197–204.
Bi X, Lv X, Liu D, Guo H, Yao G, Wang L, Liang X, Yang Y. METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther. 2021;28(3–4):335–49.
Huang H, Wang Y, Kandpal M, Zhao G, Cardenas H, Ji Y, Chaparala A, Tanner EJ, Chen J, Davuluri RV, et al. FTO-dependent N (6)-methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Can Res. 2020;80(16):3200–14.
Hosseini ES, Meryet-Figuiere M, Sabzalipoor H, Kashani HH, Nikzad H, Asemi Z. Dysregulated expression of long noncoding RNAs in gynecologic cancers. Mol Cancer. 2017;16(1):107.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Fazi F, Fatica A. Interplay between N (6)-methyladenosine (m(6)A) and non-coding RNAs in cell development and cancer. Front Cell Develop Biol. 2019;7:116.
Zhang X, Hamblin MH, Yin KJ. The long noncoding RNA Malat 1: Its physiological and pathophysiological functions. RNA Biol. 2017;14(12):1705–14.
Chen Y, Lin Y, Shu Y, He J, Gao W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol Cancer. 2020;19(1):94.
Brown JA, Kinzig CG, DeGregorio SJ, Steitz JA. Methyltransferase-like protein 16 binds the 3’-terminal triple helix of MALAT1 long noncoding RNA. Proc Natl Acad Sci USA. 2016;113(49):14013–8.
Zheng ZQ, Li ZX, Zhou GQ, Lin L, Zhang LL, Lv JW, Huang XD, Liu RQ, Chen F, He XJ, et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and upregulate ITGB3. Can Res. 2019;79(18):4612–26.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, Cao M, Cai J, Wu J, Wang X. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, Li H, Qiu M, Xu Y, **a W, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112–20.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22(9):1760–74.
Wang J, Shen C, Dong D, Zhong X, Wang Y, Yang X. Identification and verification of an immune-related lncRNA signature for predicting the prognosis of patients with bladder cancer. International immunopharmacology. 2021;90:107146.
Yuan CL, Jiang XM, Yi Y. E JF, Zhang ND, Luo X, Zou N, Wei W, Liu YY: Identification of differentially expressed lncRNAs and mRNAs in luminal-B breast cancer by RNA-sequencing. BMC Cancer. 2019;19(1):1171.
Shu X, Shu S, Cheng H. A novel lncRNA-mediated trans-regulatory mechanism in the development of cleft palate in mouse. Mol Genetics Genomic Med. 2019;7(2):00522.
Braga EA, Fridman MV, Moscovtsev AA, Filippova EA, Dmitriev AA, Kushlinskii NE: LncRNAs in Ovarian Cancer Progression, Metastasis, and Main Pathways: ceRNA and Alternative Mechanisms. Int J Mol Sci. 2020. 21(22).
Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16(4):385–95.
Iasonos A, Schrag D, Raj GV, Panageas KS. How to build and interpret a nomogram for cancer prognosis. J Clin Oncol. 2008;26(8):1364–70.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Tang Y, Li C, Zhang YJ, Wu ZH. Ferroptosis-Related Long Non-Coding RNA signature predicts the prognosis of Head and neck squamous cell carcinoma. Int J Biol Sci. 2021;17(3):702–11.
Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220.
Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang P, Liu XS. Large-scale public data reuse to model immunotherapy response and resistance. Genome medicine. 2020;12(1):21.
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):955–61.
Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PloS One. 2014;9(9):107468.
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, Li J, An P, Lu L, Luo N, et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18(1):87.
Yang X, Zhang S, He C, Xue P, Zhang L, He Z, Zang L, Feng B, Sun J, Zheng M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19(1):46.
Hu X, Peng WX, Zhou H, Jiang J, Zhou X, Huang D, Mo YY, Yang L. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2020;27(6):1782–94.
Sun T, Wu Z, Wang X, Wang Y, Hu X, Qin W, Lu S, Xu D, Wu Y, Chen Q, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39(31):5358–72.
Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou A, Liu J, Che L, Li J. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol Cancer. 2019;18(1):143.
Wang Y, Lu JH, Wu QN, ** Y, Wang DS, Chen YX, Liu J, Luo XJ, Meng Q, Pu HY, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18(1):174.
Ban Y, Tan P, Cai J, Li J, Hu M, Zhou Y, Mei Y, Tan Y, Li X, Zeng Z, et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(6):1282–96.
Rong B, Zhang Q, Wan J, **ng S, Dai R, Li Y, Cai J, **e J, Song Y, Chen J, et al. Ribosome 18S m(6)A Methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell reports. 2020;33(12):108544.
Huang X, Zhang H, Guo X, Zhu Z, Cai H, Kong X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol Oncol. 2018;11(1):88.
Yoneda R, Ueda N, Uranishi K, Hirasaki M, Kurokawa R. Long noncoding RNA pncRNA-D reduces cyclin D1 gene expression and arrests cell cycle through RNA m(6)A modification. J Biol Chem. 2020;295(17):5626–39.
Cheng F, Liu J, Zhang Y, You Q, Chen B, Cheng J, Deng C. Long non-coding RNA UBA6-AS1 promotes the malignant properties of glioblastoma by competitively binding to microRNA-760 and enhancing homeobox A2 expression. Cancer Manag Res. 2021;13:379–92.
Yang J, Li C, Li H. E C: LncRNA CACNA1G-AS1 facilitates hepatocellular carcinoma progression through the miR-2392/C1orf61 pathway. J Cell Physiol. 2019;234(10):18415–22.
Wei LJ, Bai DM, Wang ZY, Liu BC. Upregulated lncRNA CACNA1G-AS1 aggravates the progression of colorectal cancer by downregulating p53. Eur Rev Med Pharmacol Sci. 2020;24(1):130–6.
Yu PF, Kang AR, **g LJ, Wang YM. Long non-coding RNA CACNA1G-AS1 promotes cell migration, invasion and epithelial-mesenchymal transition by HNRNPA2B1 in non-small cell lung cancer. Eur Rev Med Pharmacol Sci. 2018;22(4):993–1002.
Acknowledgements
We are grateful for the data from The Cancer Genome Atlas.
Funding
This work was supported by the Research Fund of China National Health Commission (Grant Number WKJ-ZJ-2010), Medical Scientific Research Foundation of Zhejiang Province (Grant Number 2019KY495), National Natural Science Foundation of China (Grant Number 31900922), and WU JIEPING MEDICAL FOUNDATION (Grant Number 320.6750.2020–04-13).
Author information
Authors and Affiliations
Contributions
**yi Tong conceived, designed, and supervised the study. Jianfeng Zheng performed data analysis and drafted the manuscript. Ying Zhou, Jialu Guo and Benben Cao collected the data. All authors reviewed and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Hangzhou First People's Hospital (Ethical Review No.2018–147-01). Informed consent was obtained from all subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Trans-regulation lncRNA-TF-mRNA.
Additional file 2: Figure S1.
The K-M survival curve of Stage II based on the risk model. The p value was indistinctive.
Additional file 3: Table S2.
Difference in Immune microenvironment.
Additional file 4: Table S3.
Drugs with significant difference in sensitivity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, J., Guo, J., Cao, B. et al. Identification and validation of lncRNAs involved in m6A regulation for patients with ovarian cancer. Cancer Cell Int 21, 363 (2021). https://doi.org/10.1186/s12935-021-02076-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02076-7