
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive scarring interstitial lung disease with an unknown cause. Some patients may experience acute exacerbations (AE), which result in severe lung damage visible on imaging or through examination of tissue samples, often leading to high mortality rates. However, the etiology and pathogenesis of AE-IPF remain unclear. AE-IPF patients exhibit diffuse lung damage, apoptosis of type II alveolar epithelial cells, and an excessive inflammatory response. Establishing a reliable animal model of AE is critical for investigating the pathogenesis. Recent studies have reported a variety of animal models for AE-IPF, each with its own advantages and disadvantages. These models are usually established in mice with bleomycin-induced pulmonary fibrosis, using viruses, bacteria, small peptides, or specific drugs. In this review, we present an overview of different AE models, ho** to provide a useful resource for exploring the mechanisms and targeted therapies for AE-IPF.
Similar content being viewed by others
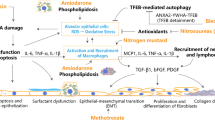


Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, fibrosing interstitial pneumonia of unknown cause that is associated with radiological and histologic features of usual interstitial pneumonia [1]. This condition leads to a decline in physiological function, severe respiratory symptoms, and ultimately fatal clinical outcomes [2,3,4]. After excluding interstitial lung diseases with known etiologies, high-resolution computed tomography findings in usual interstitial pneumonia often exhibit subpleural and basal-predominant honeycombing, traction bronchiectasis, and traction bronchiolectasis [5]. These findings may coexist with ground-glass opacification and fine reticulation, which are characteristic of IPF [5]. Histopathological features of usual interstitial pneumonia include: (1) patchy dense fibrosis with architectural distortion (i.e., destructive scarring and/or honeycombing); (2) a predilection for subpleural and paraseptal lung parenchyma; (3) fibroblast foci; and (4) the absence of features that suggest an alternative diagnosis [1]. The median overall survival time of IPF patients is only 2–3 years, which is inferior to that of some cancers [6]. Moreover, a subset of patients may experience acute exacerbations (AE) of IPF (AE-IPF), which are characterized by episodic acute declines in lung function, symptoms, and quality of life [7]. Currently, pirfenidone and nintedanib are the only approved anti-fibrotic drugs used for treating IPF [8,9,10]. Nevertheless, these two drugs can only delay the progression of the disease and are unable to completely reverse pulmonary fibrosis [10].
To date, there is no proven effective therapy for AE-IPF [11]. A recent retrospective study of 461 patients with IPF showed that 20.8% of patients developed AE and 17.7% experienced multiple AE episodes over a median follow-up of 22.9 months [12]. A meta-analysis of six clinical trials found that the incidence of AE-IPF was 4.1% per year [13]. A Japanese cohort study revealed that the probability of AE occurrence in IPF was 8.6% at one year, 12.6% at two years, and 23.9% at three years [14, 15]. The median survival time of AE-IPF cases, without undergoing lung transplantation, was only 3–4 months [16, 17]. Notably, several factors have been consistently associated with an increased risk of AE-IPF, including the presence of pathologic fibroblastic foci and lower percentage of diffusing capacity of the lungs for carbon monoxide [18, 19]. Furthermore, elevated levels of growth differentiation factor-15, leptin, lactate dehydrogenase, Krebs von den Lungen-6, heat shock proteins, and C-reactive protein in serum have been identified as highly sensitive predictors of the incidence and poor prognosis of AE-IPF [20,21,22,23,24,25,26,27]. However, the etiology and pathogenesis of AE-IPF remain unclear, and there are no effective pharmacological treatments available except lung transplantation.
Therefore, exploring the pathogenesis and searching for therapeutic targets of AE-IPF are urgent tasks. To achieve this, reliable animal models are required. Nevertheless, while numerous compounds have demonstrated efficacy in limiting the development of pulmonary fibrosis in animal models, only a few of them have managed to replicate these beneficial effects in clinical trials [16]. It is still challenging to establish a reasonable and feasible animal model. To our knowledge, there is a lack of comprehensive summaries on animal models of acute exacerbations of pulmonary fibrosis (AE-PF), which prompted us to conduct a comprehensive analysis of recent studies on animal models of AE-PF.
The pathogenesis of IPF
The development of IPF is a multifaceted process that involves intricate interactions between various cell types, such as type II alveolar epithelial cells (AEC2), fibroblasts, myofibroblasts, macrophages, neutrophils, and endothelial cells, as well as signaling pathways [28,29,30,31,99]. To generate Slc39a8 deletion mice, Slc39a8 floxed mice were crossbred with surfactant protein-C-CreER;Rosa26-tdTomato mice. Slc39a8 deletion mice exhibit a deficiency in the specific zinc transporter SLC39A8 within AEC2, resulting in spontaneous lung fibrosis [100]. Constructed a targeting vector with a mutation in surfactant protein A1 or put a mutation in surfactant protein A1 in the CRISPR/Cas9 to established surfactant protein A1-knock in mice, the homozygous mutant mice spontaneously developed pulmonary fibrosis [101].
There have also been reported several other fibrotic challenging agents in animal models and can cause varying degrees of lung fibrosis [70, 102, 103], such as asbestos (a single intratracheal administration), intratracheal instillation of particulate matter (PM 2.5) in mice [104], unilateral ureteral obstruction in rats [105], and chest radiation exposure [106, 107]. In addition, cyclophosphamide, methotrexate, 5‑fluorouracil, etc. have also been used to induce pulmonary fibrosis [108]. γHV-68 (1 × 105 PFU) were used to induce fibrosis in aged mice (≥ 15–18 months of age) and the adenovirus injected to the trachea can cause BLM-like pulmonary fibrosis [109,110,111]. The rat model of lung fibrosis was established by administration amiodarone (30 mg/kg, dissolved in 0.85% normal saline, with 0.05% Tween 80) once daily by oral gavage for 3 months [112]. Nitrogen mustard was injected intratracheally into the mice by Pavel et al., and both doses (0.625 and 0.312 mg/kg, respectively) were used to induced pulmonary fibrosis in mice [113]. Moreover, a single oral gavage or intraperitoneal injection of paraquat can induce pulmonary fibrosis in rats and mice, respectively [73, 114].
Animal models of AE-PF
Animal models of AE-PF induced by viruses
Accumulating evidence suggests that chronic viral infections may act as persistent irritant antigens or synergistic factors in susceptible hosts to induce pulmonary fibrosis [115]. The viral RNA sequencing analysis of nasopharyngeal swabs from patients with stable IPF and AE-IPF indicated that there was a significantly higher viral positive rate in AE-IPF (60%) than in patients with stable IPF (43%). The results suggested that viral infection may be a predisposed factor for AE-IPF [49]. Hence, establishing an animal model of virus-induced AE-PF would be both reasonable and feasible.
Herpes simplex virus 1 (HSV1) model
In this model, HSV1 (5 × 105 PFU) was injected intranasally to BLM mice at Day 21 and the mice were sacrificed at Day 28 [64]. Compared to BLM animals, both the BLM and HSV1 groups exhibited higher acute lung injury scores, reduced survival rates, more severe impairment in lung function, and an excessive inflammatory response, as indicated by the increased proportion of inflammatory cells and inflammatory cytokines in bronchoalveolar lavage fluid [64]. The advantage of this model is the use of human HSV1, which can easily infect humans. Moreover, this model effectively recapitulated the progressive decline of lung function and heightened mortality rates observed in patients with AE-IPF, as well as the hallmark pathological features of acute lung injury and aberrant inflammatory signaling pathways. However, the elevation of IL-17 A and endoplasmic reticulum stress induced by HSV1 infection play a role in acute lung injury and may not be the driver of pulmonary fibrosis in mice at day 28 after BLM treatment.
AE mouse model can also be established by intranasally HSV1 (5 × 105 PFU) administration on 14th day after BLM treatment [65]. The histological changes showed that the collagen deposition in the lung of BLM mice were exacerbated due to HSV1 infection. acute lung injury score, fibrosis score in mice with BLM-induced pulmonary fibrosis were increased, and the inflammatory markers such as IL-6, TNF-α and monocyte chemoattractant protein-1 in bronchoalveolar lavage fluid were also increased, while the anti-inflammatory cytokine IL-10 was decreased. Diffuse alveolar damage was characterized by interstitial edema, alveolar hemorrhage, alveolar epithelial exfoliation, and hyaline membrane formation occurred. Meanwhile, there was a reduction in forced vital capacity and lung compliance [65]. All of these findings suggested that the AEs were occurred in BLM induced pulmonary fibrosis models [65]. While the main advantage of this model was its ability to simulate some of the clinical manifestations, pathological features, and inflammatory mechanisms in AE-IPF patients, the authors did not show the hydroxyproline content, and there may be biases in the acute lung injury score and fibrosis score. Furthermore, the study did not clarify the causal relationship between endoplasmic reticulum stress and the defect of stimulator of interferon genes in AE-PF [65].
Above all, these studies demonstrated that HSV1 can induce an AE phenotype in animals with BLM-induced pulmonary fibrosis.
γ-Herpesvirus − 68 (γHV-68) model
In order to establish AE animal model, the mice were injected murine γHV-68 (5 × 104 PFU) by intranasal administration after FITC treatment for 14 days [44]. Lungs were harvested on Day 21, the excessive inflammation and collagen deposition were observed on the lung histopathology. Total lung capacity, vital capacity, and lung compliance were measured on Day 21, and the results showed that γHV-68 infection caused significant impairment of lung function in FITC-treated mice. Additionally, there was evidence of interstitial edema, intra-alveolar hemorrhage, alveolar epithelial denudation, and sloughing off of injured/dead epithelial cells [44]. The mice were treated intranasally on Day 14 with inactivated γHV-68 by exposure to ultraviolet light, on Day 21, the lungs samples were harvested, and collagen content was determined, however, there was no significant difference between mock infection and ultraviolet-inactivated γHV-68 infection. Thus, it suggested that lytic replication of the virus, not just the viral antigen, was required for the virus to aggravate FITC-induced fibrosis. While this model was able to replicate some of the clinical manifestations and pulmonary pathological changes seen in patients with AE-IPF, the mortality observed in AE mice did not increase during the 14-day period following virus treatment, unlike the high mortality of nearly 50% within a month observed in patients with AE-IPF [116]. This difference may be attributed to variations in genetics and the presence of coinfections between the animal model and the human condition. In a study by Ashley SL et al. [62] mice were treated with γHV-68 (5 × 104 PFU) intranasally at 14th day in the BLM model. At Day 21, the hydroxyproline contents of lung tissues in both BLM and γHV-68 treated mice was significantly increased compared with BLM treated mice. The results of pathology showed more severe areas of excessive inflammation reactions and interstitial collagen deposition. The evidence of apoptosis of AEC2 was observed by cleaved poly-ADP ribose polymerase [62]. The main strength of this model was its ability to elucidate the underlying mechanism by which increased apoptosis of AEC2 triggers the onset of AE, a key feature of IPF. Both studies suggested that γHV-68 virus can establish AE models in BLM animals.
Animal models of AE-PF induced by bacteria
The study showed that the lung bacterial loads in patients with AE-IPF were 4 times higher than in stable subjects. This suggests that alterations in lung bacterial loads could contribute to an increased level of lung fibrosis and the development of AE [45, 117]. As a result, bacteria are also commonly used as the triggers in animal models of AE-PF.
Staphylococcus model
In staphylococcus models [59, 60], the combined antibiotics were used in TGF-β1 transgenic mice with pulmonary fibrosis for 4 days to eliminate the chances of pulmonary infections. On the 5th day, Staphylococcus nepalensis CNDG strain (1 × 108 CFU) was injected intratracheally, and the mice were sacrificed 2 days later. Chest computed tomography (CT) and hydroxyproline of mice treated with the Staphylococcus nepalensis CNDG strain showed a significant increase in lung fibrosis and a significant neutrophil infiltration, compared with control mice treated with Staphylococcus epidermium. The increased apoptosis of AEC2 indicated that the model of Staphylococcus nepalensis CNDG strain for AE-PF was successful and feasible. In addition, TGF-β1 transgenic mice were treated with corisin, shed by Staphylococcus nepalensis CNDG, by intra-tracheal route once daily for two days before euthanasia on day 3. Compared to control mice, significantly increased infiltration of macrophages, lymphocytes and neutrophils, increased collagen deposition and concentration of inflammatory cytokines and chemokines, and enhanced apoptosis of AEC2 in the lungs were observed. This study employed a model to investigate the role of corisin, a pro-apoptotic peptide, in the development of AE in pulmonary fibrosis, and to explore its underlying molecular mechanism. Specifically, the researchers aimed to identify how corisin contributes to the onset of AE in IPF patients with bacterial infections. The study included validation experiments in both male and female mice. We eagerly anticipate the results of future studies that will build on these findings and help shed light on the complex pathogenesis of AE-IPF. While the administration of these transgenes can induce ongoing and progressive fibrosis in mice, it is important to acknowledge certain limitations of the bacterial model. These limitations include potential immune responses in the animals due to the viral vector and the expression of transgenes at levels significantly higher than what is physiologically observed [70].
Streptococcus pneumoniae model
This AE model of mice was established by intranasally infusion with Spn strains (1 × 105 CFU) at 14th day after BLM administration, compared with those treated with controls (BLM or Spn alone), the AE mice showed more severe pulmonary fibrosis and collagen deposition when sacrificed on Day 17, and the morbidity and mortality of the mice showed a dose dependence of Spn [63]. Western blot analysis showed that AIM2, activated caspase-1, and cleaved IL-1β in lung tissues were significantly increased after treatment with BLM and Spn [63]. As determined by enzyme-linked immunosorbent assay, the contents of IL-1β and IL-18 in lung tissue were significantly increased in AE mice [63]. This study revealed that activation of the AIM2 inflammasome serves as a crucial connection between glucose transporter 1-mediated glycolysis and the exacerbation of lung fibrosis induced by bacterial infection. However, the study did not report the levels of hydroxyproline in the lung tissues.
Adenovirus vector was utilized to deliver TGF-β1 to establish the model of pulmonary fibrosis in mice, and then injected Spn (1 × 107 CFU) through oral-endotracheal route at 14th day [37, 61, 118]. It was proved that the intervention can successfully induce AE by observing the collagen deposition, morphological changes and Th1/Th2 cytokine levels in lung tissue. Moreover, the depleting Treg cells in the lung could promote significantly AE-PF induced by Spn. Knippenberg S et al. [61] established another mouse model which consists of diphtheria toxin administration to transgenic surfactant protein C-diphteria toxin receptor mice to cause repetitive AEC2 injury and subsequent pulmonary fibrosis, and two weeks later, the mice were treated with Spn, the content of hydroxyproline in the lung tissue of AE mice was significantly higher than that in the control mice receiving the mock infection when they were sacrificed on Day 21. The remarkable aspect of these animal models is their capacity to mimic the exaggerated inflammatory mechanisms seen in AE-IPF.
Haemophilus influenzae model
The AE animal model with Haemophilus influenzae was also establish in BLM induced pulmonary fibrosis mice [119]. The authors administered 1 × 107 CFU non-typeable Haemophilus influenzae strain NT127 intranasally to the mice on the 7th day after BLM treatment. Then the survival rate, body weight, the bacterial burden in the lung tissues and bronchoalveolar lavage fluids on the 21th day were evaluated. The results showed that the body weight and survival of mice in both BLM and NT127 group significantly decreased compared with BLM group, while the bacterial burden in the lung tissue and bronchoalveolar lavage fluids increased. The mice in both BLM and NT127 group showed more inflammatory cell infiltration in the lung tissue and severe airway structural damage than BLM group and NT127 group. This is the first report that Gram-negative bacteria have been used to induce AE model of pulmonary fibrosis. It is believed that IL-17 is secreted from γδT cells and CD4+ T cells, and then aggravates lung injury by the recruitment of neutrophils and eosinophils in AE model mice. While the study demonstrated increased airway inflammation in the AE group of mice, the authors did not present compelling evidence of a worsening of pulmonary interstitial fibrosis. Moreover, administering NT127 on the 7th day after BLM treatment may have been considered too early.
Other models of AE-PF
Except for virus and bacteria were employed to induce AE occurrence in AE animal models, some biological or chemical agents such as LPS, repeated administration of BLM, cadmium chloride, and Nickel ions have been used in AE models.
LPS has been employed to induce acute lung injury in various preclinical models [120,121,122]. Since acute lung injury can be observed in the histopathology of patients with AE-IPF, efforts have been made to use LPS to induce AE [116]. Miyamoto H et al. [67] reported that LPS (0.05 or 0.15 mg/kg) was administered intratracheally 7 days after BLM intratracheally treated in Wistar rats. Compared with the controls, chest CT of rats in combined BLM and LPS group showed more infiltrating opacities resembling as acute respiratory distress syndrome. The histological changes showed more severe pulmonary fibrosis, and oxygen partial pressure of arterial blood decreased significantly. The plasma nitrite/nitrate, inducible nitric oxide synthase protein and mRNA levels and lung nitrotyrosine levels and the TNF-α level in bronchoalveolar lavage fluid were increased, which were similar to the pathophysiological changes of patients with AE-IPF. However, the limitations of this study are that LPS was administered on day 7, which is in the inflammatory stage rather than the fibrotic stage, and relevant indicators were monitored 24 h after LPS administration, which are different from the clinical settings. Jia K et al. [123] administered BLM intratracheal to mice on Day 1, and then LPS (1 mg/kg) was administered intratracheal on day 5, 7, and 9 to simulated AE-PF in mice. The mice in combined LPS and BLM group showed more severe inflammatory cell infiltration, thickened interalveolar space, a few collapsed alveoli in the lung tissue, a remarkable increase in collagen deposition and more severe distortion of lung architecture. Functionally, lung elastance increased and compliance and inspiratory capacity decreased. The expressions of hydroxyproline and fibrotic markers, including collagen I and α-SMA, were significantly up-regulated in mice with both BLM and LPS intervention. In particular, the expressions of inflammatory markers including IL-6, TNF-α, and IL-1β markedly increased in both BLM and LPS group compared to control group. The other study reported that LPS (0.5 mg/kg) was administered via oropharyngeal aspiration on Day 7 after BLM aspiration [124]. The changes of histological changes and CT images in the lung, lung water contents, oxygen partial pressure of arterial blood, cell counts and inflammatory cytokine levels in bronchoalveolar lavage fluid were assessed on Day 8. In both BLM and LPS group, chest CT showed diffuse ground-glass opacities, and oxygen partial pressure of arterial blood was significantly decreased. Both the BLM and LPS groups exhibited a severe inflammatory response, as evidenced by the presence of infiltrating cells in the lung histopathology and an increase in lung water content. Total cell and neutrophil counts and levels of cytokines such as monocyte chemoattractant protein-1, IL-6 and keratinocyte chemoattractant in the bronchoalveolar lavage fluid were significantly elevated in combined BLM and LPS group [124]. These models used LPS to induce AE have the advantages of excessive inflammatory responses in the lung, but lack the evidence of excessive apoptosis in AEC2. Furthermore, a single or repeated uses of LPS in the early stage is not recommended by the expert panel [16].
Repeated administration of BLM also was reported to trigger AE-PF in mice [69, 125]. The animal was injected intratracheally with BLM (4 mg/kg) to induce pulmonary fibrosis firstly, the second dose of BLM (4 mg/kg) was given on the 21th day. On Day 35, the mortality of mice and IL-6 level in bronchoalveolar lavage fluids in the two doses of BLM group were significantly higher than those in the single dose (control) group. In the AE group, the pathological changes were significantly more severe than in the control group, including the formation of hyaline membranes and the accumulation of fluid in the lungs, which are characteristic of acute lung injury in humans. The collagen scores and hydroxyproline contents in the lung and TGF-β1 level in bronchoalveolar lavage fluid were significantly higher in two doses mice than those in single-dose group. It is indicated that two doses of BLM could lead to AE of lung fibrosis. Similarly, AE model of pulmonary fibrosis was also successfully induced by a second perfusion of BLM in rats [126]. Yegen et al. established a mouse fibrosis model with four initial intratracheal injections of 0.8 UI/g BLM, given every two weeks, followed by two additional treatments of 1.6 UI/g BLM at two-week intervals to induce AE of lung fibrosis [127]. The AE group exhibited decreased body weight, higher mortality, reduced lung compliance, and significantly increased collagen synthesis compared to the BLM group. These results strongly support the successful establishment of the AE pulmonary fibrosis model [127]. An advantage of these models is their ability to corroborate the gradual progression of the disease as observed in human patients, with repeated injury and fibrosis [16]. However, the underlying assumption that fibrosis results from an external insult rather than an internal genetic/epigenetic response to injury remains unchanged [16]. Nevertheless, these models provide valuable tools for studying AE occurring in clinical settings due to non-infectious factors.
Kim MS et al. [128] established pulmonary fibrosis model in mice by tracheal instillation of polyhmethylene guanidine (PHMG, 18 µg/50µL), followed by tracheal injection of cadmium chloride (CdCl2, 0.2 µg/50µL) on days 3, 6, 9 and 12, respectively. Compared with PHMG group, the body weight of mice in PHMG and CdCl2 group was significantly reduced, the number of total cells, macrophages, neutrophils and lymphocytes in bronchoalveolar lavage fluids was increased, and collagen deposition was also increased. It was believed that PHMG and CdCl2 could induce the development AE-PF in mice. However, it should be noted that PHMG is not the optimal agent for inducing pulmonary fibrosis, and the timing and frequency of CdCl2 administration in this model were both too early and too frequent. Furthermore, the use of chemical reagents in this model is associated with high toxicity, rendering it unsuitable for widespread use.
The expressions of matrix metalloproteinase 9 and collagen I, inflammatory cells, the levels of IL-1β, TGF-β and TNF-α in bronchoalveolar lavage fluids were increased, while IL-10 level was decreased in BLM and nickel chloride mice compared with BLM group [129]. The hydroxyproline contents and fibrosis scores were also increased in BLM and nickel chloride group than in BLM group [129]. However, it should be noted that the simultaneous administration of nickel chloride and BLM on Day 0 in this model is not consistent with clinical practice. BLM-treated peroxiredoxin 4-transgenic mice showed more severe collagen deposition and strong immunostaining of fibronectin and worse prognosis compared to BLM-treated mice, chest CT demonstrated traction bronchiectasis and severe consolidations [130]. This finding may offer valuable insights for patients with genetically related AE-IPF.
The AE-PF animal models have provided valuable insights into disease mechanisms, but they also have certain limitations that must be addressed. While existing models can partially replicate AE features, further refinement is necessary to fully capture the complexity of this condition. To achieve a comprehensive understanding of how cell-cell interactions and soluble mediators affect AE, both biochemical and histologic analyses are crucial.
The BLM-induced mouse models produce stable and reproducible fibrotic characteristics, but it cannot mimic the progressive and irreversible nature of IPF. Recently, a strategy involving repeated BLM instillations had resulted in progressive fibrosis, recapitulating the histological and imaging characteristics of IPF. This approach offers insights into addressing this issue [82]. On the other hand, the FITC-induced lung fibrosis model accurately displays lesions through fluorescence signals, but lack of clinical relevance and inability to form fibroblastic foci limit its use. In addition, the TGF-β transgenic model induces sufficient fibrosis, but overexpression of transgenes may result in excessive fibrosis, and mice may develop an immune response to adenoviral vectors. Simulating the pathogenesis of IPF by damaging AEC2 is a cumbersome process, and repeated drug use may lead to drug resistance. Moreover, the toxic nature of reagents used in this approach poses risks to laboratory personnel. Although using viruses or bacteria to induce AE can accurately simulate clinical settings, some pathogens may only exacerbate inflammation and not fibrosis. Chemical reagents can be used to induce AE in mouse models, but timing and frequency of administration must be optimized, and the toxicity of certain reagents limits their widespread use. Despite these limitations, animal models provide a valuable tool for understanding the pathogenesis of AE-IPF and identifying potential therapeutic targets. With continued refinement and validation, these models may offer a promising avenue for advancing our understanding of this devastating disease.
Taking into account the advantages and disadvantages of established animal models, researchers are actively exploring the development of novel animal models. Pigs, due to their anatomical size and structure, physiology, immunology, and genome similarity to humans, have been utilized in the investigation of various lung diseases [131, 132]. A model of unilateral acute lung injury in pigs has been established, validated by histopathology and an inflammatory response [133]. This model can serve as a basis for comparing the pathophysiological changes in both the injured and uninjured lungs within the same animal [133], and holds potential for the study of AE-IPF in the future. Numerous studies have explored the utilization of sheep as an animal model for pulmonary fibrosis. Organ et al. employed a bronchoscope to inject BLM into the lung base, effectively replicating the clinical characteristics of IPF, which often manifest as subpleural and basal-predominant [134]. The study demonstrated a correlation between fibrosis and impaired lung function for up to seven weeks following BLM treatment [134]. Leveraging the larger size of sheep, multiple blood gas analyses were conducted, facilitating the monitoring of oxygen and carbon dioxide partial pressures [135]. This approach accurately simulates clinical practice for IPF patients who experience severe hypoxia and necessitate dynamic blood gas monitoring. In addition, Perera et al. successfully reproduced factors that may be involved in the pathogenesis of IPF, such as ER stress and AEC2 and macrophage apoptosis [136]. Importantly, their research indicated that administering calcium-activated potassium channel inhibitors in vivo could alleviate endoplasmic reticulum stress and the apoptosis of AEC2 and macrophages [136]. Furthermore, significant miRNA expression similarities between sheep models and IPF patients were revealed by database analysis, in contrast to mice [137]. This valuable information contributes significantly to the study of IPF pathogenesis and the quest for therapeutic targets.
Furthermore, since chest imaging and pulmonary function indicators are often the primary endpoint in clinical trials [70], these parameters warrant further investigation in future animal experiments. In upcoming investigations, some novel peripheral blood biomarkers may have wider applications. Among these are particular sections of type IV collagen, such as a1 (C4M12a1) and a3 (C4M12a3) chains, as well as neoantigens generated by the cleavage of matrix metalloproteinases [16].
Lung slice cultures offer several benefits, including the identification of new targets, the facilitation of kinetic investigations, the comparison of biological processes between human and murine systems, and the examination of slices from IPF-affected lungs [16]. However, precision cut lung tissues have a significant limitation in that they have a short lifespan of approximately one week, which makes them unsuitable for use as chronic, long-term drug testing platforms [138].
We summarize the currently published AE animal models of pulmonary fibrosis in Table 1. Based on the data, there is considerable inconsistency in time points of triggers to induce AE. As we know, the administration of BLM is the commonly used experimental system to study IPF pathogenesis [103, 139]. This dosing regimen transforms into a fibrotic response from approximately day 14, and may spontaneously resolve beyond 28 days [70]. For an accurate evaluation of anti-fibrotic efficacy, the intervention should target fibrosis [139]. Similarly, to accurately assess the effect of challenging agents for AE and study pathogenesis of AE-PF, we propose an ideal AE model should be performed in the fibrotic phase (on the 14th day after BLM administration) and collect the samples of blood, bronchoalveolar lavage fluid and lung tissues at Day 21 to evaluate whether AE occurred and the possible mechanism (shown in Fig. 2).

Schematic diagram depicting the mouse model of AE-PF
On Day 0, mice were administered with a single dose of BLM via the transtracheal route. On Day 14 after BLM treatment, mice in the AE group were challenged with bacteria, viruses, or other challenging reagents such as BLM or lipopolysaccharide. Chest CT scans were performed on Day 20 before animal sacrificed. The mice were killed on Day 21, and the samples of blood, bronchoalveolar lavage fluids, and lung tissues were collected
Abbreviations: AE: acute exacerbation; AE-PF: acute exacerbation of pulmonary fibrosis; BLM: bleomycin; CT: computed tomography
Conclusion
Establishing a consistent and reliable animal model of AE-PF holds great significance in understanding its pathogenesis. We have summarized the various animal types, inducers, and dosages employed in existing models, with bleomycin and mice continuing to be the most frequently utilized choices. Each model exhibits its unique attributes, and the timing of interventions plays a vital role in the development of an effective AE-PF model. Administering challenging agents for AE at the peak level of fibrosis would be the ideal approach. Although these animal models may not replicate the exact characteristics of AE-IPF patients, they nevertheless provide valuable insights into the pathobiology associated with cells, mediators, and signaling pathways that may be involved in fibrosis as well as AEs. Additionally, the AE animal model of pulmonary fibrosis serves as a crucial means to evaluate hypotheses regarding novel therapies. Hopefully, establishing an ideal animal model of AE-PF will provide an important tool for exploring the mechanisms underlying this debilitating disease and identifying potential therapeutic targets.
Data Availability
Not applicable.
Abbreviations
- AE:
-
acute exacerbation
- AE-PF:
-
acute exacerbations of pulmonary fibrosis
- AEC2:
-
type II alveolar epithelial cells
- BLM:
-
bleomycin
- FITC:
-
fluorescein isothiocyanate
- HSV1:
-
herpes simplex virus 1
- IPF:
-
idiopathic pulmonary fibrosis
- γHV-68:
-
γ-Herpesvirus − 68
- α-SMA:
-
α-smooth muscle actin
- LPS:
-
lipopolysaccharide
- Spn:
-
streptococcus pneumoniae
- TGF-β:
-
transforming growth factor β
- TNF-α:
-
tumor necrosis factor α
References
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an update) and Progressive pulmonary fibrosis in adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205:e18–47.
Podolanczuk AJ, Thomson CC, Remy-Jardin M, Richeldi L, Martinez FJ, Kolb M, et al. Idiopathic Pulmonary Fibrosis: state of the art for 2023. Eur Respir J. 2023;61:2200957.
Richeldi L, Collard HR, Jones MG. Idiopathic Pulmonary Fibrosis. The Lancet. 2017;389:1941–52.
Kolb M, Bondue B, Pesci A, Miyazaki Y, Song JW, Bhatt NY, et al. Acute exacerbations of progressive-fibrosing interstitial lung Diseases. Eur Respir Rev. 2018;27:180071.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198:e44–68.
du Bois RM. An earlier and more confident diagnosis of Idiopathic Pulmonary Fibrosis. Eur Respiratory Rev. 2012;21:141–6.
Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, et al. Acute exacerbations of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2007;176:636–43.
King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of Pirfenidone in patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370:2083–92.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370:2071–82.
Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555–66.
Khor YH. Antifibrotic therapy for Idiopathic Pulmonary Fibrosis. Chest. 2021;160:1589–91.
Bonniaud P, Fabre A, Frossard N, Guignabert C, Inman M, Kuebler WM, et al. Optimising experimental research in Respiratory Diseases: an ERS statement. Eur Respir J. 2018;51:1702133.
Atkins CP, Loke YK, Wilson AM. Outcomes in Idiopathic Pulmonary Fibrosis: a meta-analysis from placebo controlled trials. Respir Med. 2014;108:376–87.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194:265–75.
Kondoh Y, Taniguchi H, Ebina M, Azuma A, Ogura T, Taguchi Y, et al. Risk factors for acute exacerbation of Idiopathic Pulmonary Fibrosis – extended analysis of pirfenidone trial in Japan. Respiratory Invest. 2015;53:271–8.
Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, et al. An official American thoracic Society Workshop Report: Use of Animal models for the Preclinical Assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;56:667–79.
Juarez MM, Chan AL, Norris AG, Morrissey BM, Albertson TE. Acute exacerbation of Idiopathic Pulmonary fibrosis—a review of current and novel pharmacotherapies. J Thorac Disease. 2015;7:21.
Collard HR, Richeldi L, Kim DS, Taniguchi H, Tschoepe I, Luisetti M, et al. Acute exacerbations in the INPULSIS trials of nintedanib in Idiopathic Pulmonary Fibrosis. Eur Respir J. 2017;49:1601339.
Song JW, Hong S-B, Lim C-M, Koh Y, Kim DS. Acute exacerbation of Idiopathic Pulmonary Fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–63.
Kahloon RA, Xue J, Bhargava A, Csizmadia E, Otterbein L, Kass DJ, et al. Patients with Idiopathic Pulmonary Fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med. 2013;187:768–75.
Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, et al. Baseline KL-6 predicts increased risk for acute exacerbation of Idiopathic Pulmonary Fibrosis. Respir Med. 2014;108:1031–9.
Kishaba T, Nei Y, Momose M, Nagano H, Yamashiro S. Clinical characteristics based on the New Criteria of Acute Exacerbation in patients with Idiopathic Pulmonary Fibrosis. Eurasian J Med. 2018;50:6–10.
Cao M, Gu L, Guo L, Liu M, Wang T, Zhang J, et al. Elevated expression of growth differentiation Factor-15 is Associated with Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Front Immunol. 2022;13:891448.
Cao M, Swigris JJ, Wang X, Cao M, Qiu Y, Huang M, et al. Plasma leptin is elevated in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat Inflamm. 2016;2016:1–7.
Jain M, Budinger GRS, Lo A, Urich D, Rivera SE, Ghosh AK, et al. Leptin promotes fibroproliferative Acute Respiratory Distress Syndrome by inhibiting peroxisome proliferator–activated Receptor-γ. Am J Respir Crit Care Med. 2011;183:1490–8.
Tanguy J, Pommerolle L, Garrido C, Kolb M, Bonniaud P, Goirand F, et al. Extracellular heat shock proteins as therapeutic targets and biomarkers in fibrosing interstitial Lung Diseases. IJMS. 2021;22:9316.
Radwanska A, Cottage CT, Piras A, Overed-Sayer C, Sihlbom C, Budida R, et al. Increased expression and accumulation of GDF15 in IPF extracellular matrix contribute to fibrosis. JCI Insight. 2022;7:e153058.
Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol Mech Dis. 2022;17:515–46.
Yanagihara T, Sato S, Upagupta C, Kolb M. What have we learned from basic science studies on Idiopathic Pulmonary Fibrosis? Eur Respir Rev. 2019;28:190029.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63.
Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE. Baseline BAL Neutrophilia predicts early mortality in Idiopathic Pulmonary Fibrosis. Chest. 2008;133:226–32.
Cui H, **e N, Banerjee S, Ge J, Jiang D, Dey T, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol. 2021;64:115–25.
Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. N Engl J Med. 2020;383:958–68.
Yanagihara T, Chong SG, Gholiof M, Lipson KE, Zhou Q, Scallan C et al. Connective-Tissue Growth Factor (CTGF/CCN2) Contributes to TGF-β1-Induced Lung Fibrosis [Internet]. Cell Biology; 2020 Jul. https://doi.org/10.1101/2020.07.04.187492.
Liu GY, Budinger GRS, Dematte JE. Advances in the management of Idiopathic Pulmonary Fibrosis and Progressive pulmonary fibrosis. BMJ. 2022;377:e066354.
Kishore A, Petrek M. Roles of macrophage polarization and macrophage-derived miRNAs in Pulmonary Fibrosis. Front Immunol. 2021;12:678457.
Moyé S, Bormann T, Maus R, Sparwasser T, Sandrock I, Prinz I, et al. Regulatory T cells limit Pneumococcus-Induced Exacerbation of Lung Fibrosis in mice. JI. 2020;204:2429–38.
Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic Drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. 2017;50:1602367.
Yanagihara T, Scallan C, Ask K, Kolb MRJ. Emerging therapeutic targets for Idiopathic Pulmonary Fibrosis: preclinical progress and therapeutic implications. Expert Opin Ther Targets. 2021;25:939–48.
Bando M, Ohno S, Hosono T, Yanase K, Sato Y, Sohara Y, et al. Risk of Acute Exacerbation after Video-assisted thoracoscopic lung biopsy for interstitial lung Disease. J Bronchol Interventional Pulmonol. 2009;16:229–35.
Johannson KA, Vittinghoff E, Lee K, Balmes JR, Ji W, Kaplan GG, et al. Acute exacerbation of Idiopathic Pulmonary Fibrosis associated with air pollution exposure. Eur Respir J. 2014;43:1124–31.
Schupp JC, Binder H, Jäger B, Cillis G, Zissel G, Müller-Quernheim J et al. Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Ryffel B, editor. PLoS ONE. 2015;10:e0116775.
Tomos I, Dimakopoulou K, Manali ED, Papiris SA, Karakatsani A. Long-term personal air pollution exposure and risk for acute exacerbation of Idiopathic Pulmonary Fibrosis. Environ Health. 2021;20:99.
McMillan TR, Moore BB, Weinberg JB, Vannella KM, Fields WB, Christensen PJ, et al. Exacerbation of established pulmonary fibrosis in a murine model by Gammaherpesvirus. Am J Respir Crit Care Med. 2008;177:771–80.
Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WOC, et al. Changes in the respiratory microbiome during acute exacerbations of Idiopathic Pulmonary Fibrosis. Respir Res. 2017;18:29.
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, et al. Lung microbiome and Disease progression in Idiopathic Pulmonary Fibrosis: an analysis of the COMET study. The Lancet Respiratory Medicine. 2014;2:548–56.
Weng D, Chen X-Q, Qiu H, Zhang Y, Li Q-H, Zhao M-M, et al. The role of Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat Inflamm. 2019;2019:1–10.
Invernizzi R, Molyneaux PL. The contribution of Infection and the respiratory microbiome in acute exacerbations of Idiopathic Pulmonary Fibrosis. Eur Respir Rev. 2019;28:190045.
Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, et al. Viral Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2011;183:1698–702.
Saraya T, Kimura H, Kurai D, Tamura M, Ogawa Y, Mikura S, et al. Clinical significance of respiratory virus detection in patients with acute exacerbation of interstitial lung Diseases. Respir Med. 2018;136:88–92.
Ghincea A, Ryu C, Herzog EL. An Acute Exacerbation of Idiopathic Pulmonary Fibrosis after BNT162b2 mRNA COVID-19 vaccination: a Case Report. Chest. 2022;161:e71–3.
Omote N, Kanemitsu Y, Inoue T, Yonezawa T, Ichihashi T, Shindo Y, et al. Successful treatment with high-dose steroids for Acute Exacerbation of Idiopathic Pulmonary Fibrosis triggered by COVID-19. Intern Med. 2022;61:233–6.
Umeda Y, Morikawa M, Anzai M, Sumida Y, Kadowaki M, Ameshima S, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis after Pandemic Influenza A (H1N1) vaccination. Intern Med. 2010;49:2333–6.
Marron RM, Vega Sanchez ME, Clauss H, Mamary AJ. Acute Hypoxemic Respiratory Failure and Native Lung Idiopathic Pulmonary Fibrosis Exacerbation in Single-lung Transplant Patients with Cytomegalovirus Disease: A Case Series. Transplantation Proceedings. 2019;51:3391–4.
Moore BB, Moore TA. Viruses in Idiopathic Pulmonary Fibrosis. Etiology and Exacerbation Annals ATS. 2015;12:186–92.
Poletti V, Ravaglia C, Gurioli C, Piciucchi S, Dubini A, Cavazza A, et al. Invasive diagnostic techniques in idiopathic interstitial pneumonias. Respirology. 2016;21:44–50.
Iyoda A, Azuma Y, Sakamoto S, Homma S, Sano A. Surgical treatment for patients with Idiopathic Pulmonary Fibrosis and Lung cancer: postoperative acute exacerbation of Idiopathic Pulmonary Fibrosis and outcomes. Surg Today. 2022;52:736–44.
Qiu M, Chen Y, Ye Q. Risk factors for acute exacerbation of Idiopathic Pulmonary Fibrosis: a systematic review and meta-analysis. Clin Respir J. 2018;12:1084–92.
D’Alessandro-Gabazza CN, Kobayashi T, Yasuma T, Toda M, Kim H, Fujimoto H, et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat Commun. 2020;11:1539.
D’Alessandro-Gabazza CN, Yasuma T, Kobayashi T, Toda M, Abdel-Hamid AM, Fujimoto H, et al. Inhibition of lung microbiota-derived proapoptotic peptides ameliorates acute exacerbation of pulmonary fibrosis. Nat Commun. 2022;13:1558.
Knippenberg S, Ueberberg B, Maus R, Bohling J, Ding N, Tort Tarres M, et al. Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax. 2015;70:636–46.
Ashley SL, Jegal Y, Moore TA, van Dyk LF, Laouar Y, Moore BB. γ-Herpes virus-68, but not Pseudomonas aeruginosa or Influenza A (H1N1), exacerbates established murine lung fibrosis. Am J Physiology-Lung Cell Mol Physiol. 2014;307:L219–30.
Cho SJ, Moon J-S, Nikahira K, Yun HS, Harris R, Hong KS, et al. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax. 2020;75:227–36.
Chen T, Qiu H, Zhao M-M, Chen S-S, Wu Q, Zhou N-Y, et al. IL-17A contributes to HSV1 infection-induced acute lung injury in a mouse model of pulmonary fibrosis. J Cell Mol Med. 2019;23:908–19.
Qiu H, Weng D, Chen T, Shen L, Chen S-S, Wei Y-R, et al. Stimulator of Interferon genes Deficiency in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Front Immunol. 2017;8:1756.
McElroy AN, Invernizzi R, Laskowska JW, O’Neill A, Doroudian M, Moghoofei M, et al. Candidate role for toll-like receptor 3 L412F polymorphism and Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2022;205:550–62.
Miyamoto H, Takemura S, Minamiyama Y, Tsukioka T, Toda M, Nishiyama N, et al. Acute exacerbation of Idiopathic Pulmonary Fibrosis model by small amount of lipopolysaccharide in rats. J Clin Biochem Nutr. 2022;70:129–39.
Yamaguchi K, Iwamoto H, Sakamoto S, Horimasu Y, Masuda T, Miyamoto S, et al. Association of the RAGE/RAGE-ligand axis with interstitial lung Disease and its acute exacerbation. Respiratory Invest. 2022;60:531–42.
Fikry H, Saleh LA, Gawad SA. Therapeutic effect of adipose-derived mesenchymal stem cells (AD-MSCs) compared to pirfenidone on corticosteroid resistance in a mouse model of acute exacerbation of Idiopathic Pulmonary Fibrosis. Histol Histopathol. 2022;37:1065–83.
Carrington R, Jordan S, Pitchford SC, Page CP. Use of animal models in IPF research. Pulm Pharmacol Ther. 2018;51:73–8.
Larson-Casey JL, He C, Che P, Wang M, Cai G, Kim Y et al. Technical advance: The use of tree shrews as a model of pulmonary fibrosis. Zissel G, editor. PLoS ONE. 2020;15:e0241323.
Miele A, Dhaliwal K, Du Toit N, Murchison JT, Dhaliwal C, Brooks H, et al. Chronic Pleuropulmonary fibrosis and Elastosis of aged donkeys. Chest. 2014;145:1325–32.
Murray RE, Gibson JE. A comparative study of paraquat intoxication in rats, guinea pigs and monkeys. Exp Mol Pathol. 1972;17:317–25.
Laurila HP, Rajamäki MM. Update on Canine Idiopathic Pulmonary Fibrosis in West Highland White Terriers. Veterinary Clin North America: Small Anim Pract. 2020;50:431–46.
Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiology-Lung Cell Mol Physiol. 2008;294:L152–60.
Yanagihara T, Chong SG, Vierhout M, Hirota JA, Ask K, Kolb M. Current models of pulmonary fibrosis for future drug discovery efforts. Expert Opin Drug Discov. 2020;15:931–41.
Sleijfer S. Bleomycin-Induced Pneumonitis. Chest. 2001;120:617–24.
Adamson IYR, Bowden DH. The Pathogenesis of Bleomycin-Induced Pulmonary Fibrosis in mice. 1974;77:14.
Thrall RS, McCormick JR, Jack RM, McReynolds RA, Ward PA. Bleomycin-induced pulmonary fibrosis in the rat: inhibition by indomethacin. Am J Pathol. 1979;95:117–30.
Arif M, Basu A, Wolf KM, Park JK, Pommerolle L, Behee M, et al. An integrative Multiomics Framework for Identification of therapeutic targets in Pulmonary Fibrosis. Adv Sci. 2023;10:2207454.
Rittié L, editor. Fibrosis: Methods and Protocols [Internet]. New York, NY: Springer New York. ; 2017 [cited 2022 Aug 31]. Available from: http://springer.longhoe.net/https://doi.org/10.1007/978-1-4939-7113-8.
Redente EF, Black BP, Backos DS, Bahadur AN, Humphries SM, Lynch DA, et al. Persistent, Progressive pulmonary fibrosis and epithelial remodeling in mice. Am J Respir Cell Mol Biol. 2021;64:669–76.
Degryse AL, Lawson WE. Progress toward improving animal models for Idiopathic Pulmonary Fibrosis. Am J Med Sci. 2011;341:444–9.
Krefft S, Wolff J, Rose C, Silicosis. An update and guide for clinicians. Clin Chest Med. 2020;41:709–22.
Sun W, Li Y, Ma D, Liu Y, Xu Q, Cheng D, et al. ALKBH5 promotes lung fibroblast activation and silica-induced pulmonary fibrosis through miR-320a-3p and FOXM1. Cell Mol Biol Lett. 2022;27:26.
Shen J, Feng J, Wu Z, Ou Y, Zhang Q, Nong Q, et al. Apelin prevents and alleviates crystalline silica-induced pulmonary fibrosis via inhibiting transforming growth factor Beta 1-triggered fibroblast activation. Int J Biol Sci. 2023;19:4004–19.
Abdelrahman RS, Shawky NM. Trimetazidine, a metabolic modulator, attenuates silica-induced pulmonary fibrosis and decreases lactate levels and LDH activity in rats. J Biochem & Molecular Tox. 2022;36:e23071.
Ogawa T, Shichino S, Ueha S, Ogawa S, Matsushima K. Complement protein C1q activates lung fibroblasts and exacerbates silica-induced pulmonary fibrosis in mice. Biochem Biophys Res Commun. 2022;603:88–93.
Sugimoto N, Suzukawa M, Nagase H, Koizumi Y, Ro S, Kobayashi K, et al. IL-9 Blockade suppresses silica-induced lung inflammation and fibrosis in mice. Am J Respir Cell Mol Biol. 2019;60:232–43.
Liu H, Li Y, Zou Y, Zhang X, Shi X, Yin Z et al. Influence of miRNA-30a-5p on Pulmonary Fibrosis in Mice with Streptococcus pneumoniae Infection through Regulation of Autophagy by Beclin-1. Liu J, editor. BioMed Research International. 2021;2021:1–8.
Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early Growth Response Gene 1–mediated apoptosis is essential for transforming growth factor β1–induced pulmonary fibrosis. J Exp Med. 2004;200:377–89.
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194:809–21.
Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–36.
Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted Injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–63.
Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Flaherty KR, Martinez FJ, et al. Therapeutic targeting of CC Ligand 21 or CC chemokine receptor 7 abrogates Pulmonary Fibrosis Induced by the adoptive transfer of human pulmonary fibroblasts to Immunodeficient mice. Am J Pathol. 2007;170:1152–64.
Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ et al. A SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight [Internet]. 2019 [cited 2023 May 21]; Available from: http://insight.jci.org/articles/view/126125.
Venosa A, Katzen J, Tomer Y, Kopp M, Jamil S, Russo SJ, et al. Epithelial expression of an interstitial lung Disease–Associated Mutation in surfactant Protein-C modulates recruitment and activation of key myeloid cell populations in mice. J Immunol. 2019;202:2760–71.
Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. 2018;128:4008–24.
Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells Drives Progressive Pulmonary Fibrosis. Am J Respir Crit Care Med. 2021;203:707–17.
Liang J, Huang G, Liu X, Taghavifar F, Liu N, Wang Y, et al. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and Idiopathic Pulmonary Fibrosis. J Clin Invest. 2022;132:e157338.
Takezaki A, Tsukumo S, Setoguchi Y, Ledford JG, Goto H, Hosomichi K, et al. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with Idiopathic Pulmonary Fibrosis. J Exp Med. 2019;216:2724–35.
Moore B, Lawson B, Oury WE, Sisson TD, Raghavendran TH, Hogaboam K. Animal models of Fibrotic Lung Disease. Am J Respir Cell Mol Biol. 2013;49:167–79.
Mekhael O, Naiel S, Vierhout M, Hayat AI, Revill SD, Abed S et al. Mouse Models of Lung Fibrosis. In: Hinz B, Lagares D, editors. Myofibroblasts [Internet]. New York, NY: Springer US; 2021 [cited 2023 May 3]. p. 291–321. Available from: https://springer.longhoe.net/https://doi.org/10.1007/978-1-0716-1382-5_21.
Guo L, Bai S, Ding S, Zhao L, Xu S, Wang X. PM2.5 Exposure Induces Lung Injury and Fibrosis by Regulating Ferroptosis via TGF-β Signaling. Yang X, editor. Disease Markers. 2022;2022:1–11.
Yang F, Chang Y, Zhang C, **ong Y, Wang X, Ma X, et al. UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int Immunopharmacol. 2021;93:107396.
Suzuki T, Kropski JA, Chen J, Carrier EJ, Chen X, Sherrill TP, et al. Thromboxane–prostanoid receptor signaling drives persistent fibroblast activation in Pulmonary Fibrosis. Am J Respir Crit Care Med. 2022;206:596–607.
Ghita M, Dunne V, Hanna GG, Prise KM, Williams JP, Butterworth KT. Preclinical models of radiation-induced lung damage: challenges and opportunities for small animal radiotherapy. BJR. 2019;92:20180473.
Li S, Shi J, Tang H. Animal models of drug-induced pulmonary fibrosis: an overview of molecular mechanisms and characteristics. Cell Biol Toxicol. 2022;38:699–723.
Torres-González E, Bueno M, Tanaka A, Krug LT, Cheng D-S, Polosukhin VV, et al. Role of endoplasmic reticulum stress in Age-Related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol. 2012;46:748–56.
Naik PN, Horowitz JC, Moore TA, Wilke CA, Toews GB, Moore BB. Pulmonary Fibrosis Induced by -Herpesvirus in aged mice is Associated with increased fibroblast responsiveness to transforming growth Factor-. The journals of Gerontology Series A: Biological sciences and Medical sciences. 2012;67:714–25.
Zhou Q, Chen T, Bozkanat M, Ibe JCF, Christman JW, Raj JU et al. Intratracheal Instillation of High Dose Adenoviral Vectors Is Sufficient to Induce Lung Injury and Fibrosis in Mice. Su X, editor. PLoS ONE. 2014;9:e116142.
Abdel Halim AS, Ahmed HH, Aglan HA, Abdel Hamid FF, Mohamed MR. Role of bone marrow-derived mesenchymal stem cells in alleviating pulmonary epithelium damage and extracellular matrix remodeling in a rat model of lung fibrosis induced by amiodarone. Biotech Histochem. 2021;96:418–30.
Solopov P, Marinova M, Dimitropoulou C, Colunga Biancatelli RML, Catravas JD. Development of chronic lung injury and pulmonary fibrosis in mice following acute exposure to nitrogen mustard. Inhalation Toxicol. 2020;32:141–54.
Cui Y, **n H, Tao Y, Mei L, Wang Z. Arenaria kansuensis attenuates pulmonary fibrosis in mice via the activation of Nrf2 pathway and the inhibition of NF-kB / TGF ‐beta1/Smad2/3 pathway. Phytother Res. 2021;35:974–86.
Stoolman JS, Vannella KM, Coomes SM, Wilke CA, Sisson TH, Toews GB, et al. Latent Infection by γherpesvirus stimulates profibrotic mediator release from multiple cell types. Am J Physiol Lung Cell Mol Physiol. 2011;300:L274–285.
Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society Statement: update of the International Multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48.
Casado F, Morty RE. The emergence of preclinical studies on the role of the microbiome in lung development and experimental animal models of bronchopulmonary dysplasia. Am J Physiology-Lung Cell Mol Physiol. 2020;318:L402–4.
Bormann T, Maus R, Stolper J, Jonigk D, Welte T, Gauldie J, et al. Role of the COX2-PGE 2 axis in S. pneumoniae -induced exacerbation of experimental fibrosis. Am J Physiology-Lung Cell Mol Physiol. 2021;320:L377–92.
Chen S, Zhang X, Yang C, Wang S, Shen H. Essential role of IL-17 in acute exacerbation of pulmonary fibrosis induced by non-typeable Haemophilus influenzae. Theranostics. 2022;12:5125–37.
Zhou Y, ** T, Gao M, Luo Z, Mutahir S, Shi C, et al. Aqueous extract of Platycodon grandiflorus attenuates lipopolysaccharide-induced apoptosis and inflammatory cell infiltration in mouse lungs by inhibiting PI3K/Akt signaling. Chin Med. 2023;18:36.
Li J, Lu K, Sun F, Tan S, Zhang X, Sheng W, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19:96.
D’Alessio FR. Mouse Models of Acute Lung Injury and ARDS. In: Alper S, Janssen WJ, editors. Lung Innate Immunity and Inflammation: Methods and Protocols [Internet]. New York, NY: Springer; 2018 [cited 2023 Sep 29]. p. 341–50. https://doi.org/10.1007/978-1-4939-8570-8_22.
Jia K, Wu J, Li Y, Liu J, Liu R, Cai Y et al. A novel pulmonary fibrosis murine model with immune-related liver injury. Anim Models and Exp Med. 2022;ame2.12263.
Kimura T, Nojiri T, Hosoda H, Shintani Y, Inoue M, Miyazato M, et al. Exacerbation of bleomycin-induced injury by lipopolysaccharide in mice: establishment of a mouse model for acute exacerbation of interstitial lung Diseases. Eur J Cardiothorac Surg. 2015;48:e85–91.
Wei Y-R, Qiu H, Wu Q, Du Y-K, Yin Z-F, Chen S-S, et al. Establishment of the mouse model of acute exacerbation of Idiopathic Pulmonary Fibrosis. Exp Lung Res. 2016;42:75–86.
Chen S-S, Yin Z-F, Chen T, Qiu H, Wei Y-R, Du S-S, et al. Development of a non-infectious rat model of acute exacerbation of Idiopathic Pulmonary Fibrosis. J Thorac Dis. 2017;9:96–105.
Yegen C-H, Haine L, Da Costa Ferreira K, Marchant D, Bernaudin J-F, Planès C, et al. A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in mice. Cells. 2022;11:3379.
Kim M-S, Kim S-H, Jeon D, Kim H-Y, Han J-Y, Kim B, et al. Low-dose cadmium exposure exacerbates polyhexamethylene guanidine-induced lung fibrosis in mice. J Toxicol Environ Health Part A. 2018;81:384–96.
Yang L, Lin Z, Wang Y, Li C, Xu W, Li Q, et al. Nickle(II) ions exacerbate bleomycin-induced pulmonary inflammation and fibrosis by activating the ROS/Akt signaling pathway. Environ Sci Pollut Res. 2018;25:4406–18.
Hanaka T, Kido T, Noguchi S, Yamada S, Noguchi H, Guo X, et al. The overexpression of peroxiredoxin-4 affects the progression of Idiopathic Pulmonary Fibrosis. BMC Pulm Med. 2019;19:265.
Lunney JK, Van Goor A, Walker KE, Hailstock T, Franklin J, Dai C. Importance of the pig as a human biomedical model. Sci Transl Med. 2021;13:eabd5758.
Wei J, Zhang W, Li J, ** Y, Qiu Z. Application of the transgenic pig model in biomedical research: a review. Front Cell Dev Biol. 2022;10:1031812.
Geilen J, Kainz M, Zapletal B, Geleff S, Wisser W, Bohle B, et al. Unilateral acute lung injury in pig: a promising animal model. J Transl Med. 2022;20:548.
Organ L, Bacci B, Koumoundouros E, Barcham G, Milne M, Kimpton W, et al. Structural and functional correlations in a large animal model of bleomycin-induced pulmonary fibrosis. BMC Pulm Med. 2015;15:81.
Shinozaki S, Kobayashi T, Kubo K, Sekiguchi M. Pulmonary hemodynamics and lung function during chronic paraquat Poisoning in Sheep: possible role of reactive oxygen species. Am Rev Respir Dis. 1992;146:775–80.
Perera UE, Organ L, Dewage SNV, Derseh HB, Stent A, Snibson KJ. Increased Levels of ER Stress and Apoptosis in a Sheep Model for Pulmonary Fibrosis Are Alleviated by In Vivo Blockade of the KCa3.1 Ion Channel. Santus P, editor. Canadian Respiratory Journal. 2021;2021:1–11.
Perera UE, Derseh HB, Dewage SNV, Stent A, Wijayarathna R, Snibson KJ. Evaluation of microRNA expression in a sheep model for lung fibrosis. BMC Genomics. 2021;22:827.
Sundarakrishnan A, Chen Y, Black LD, Aldridge BB, Kaplan DL. Engineered cell and tissue models of pulmonary fibrosis. Adv Drug Deliv Rev. 2018;129:78–94.
Kolb P, Upagupta C, Vierhout M, Ayaub E, Bellaye PS, Gauldie J, et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J. 2020;55:1901105.
Acknowledgements
We are greatly appreciated for Prof. Dianhua Jiang (Cedars-Sinai Medical Center, Los Angeles, USA) for reviewing and providing valuable comments on our manuscript.
Funding
This work was supported by National Natural Science Foundation of China (82070064, 81670059 and 81200049), Fundings for Clinical Trials from the Affiliated Drum Tower Hospital, Medical School of Nan**g University (2022-LCYJ-MS-11).
Author information
Authors and Affiliations
Contributions
Xu Ye, Mingrui Zhang and Huimin Gu wrote the main manuscript text and Mengying Liu prepared Figs. 1 and 2; Table 1. Yichao Zhao, Yanchen Shi, Shufei Wu, Cheng Jiang, **aoling Ye, Huihui Zhu, Qi Li collected the data. **nmei Huang was responsible for revising and reviewing the manuscript. Mengshu Cao designed the manuscript and helped revise and review and finalize the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ye, X., Zhang, M., Gu, H. et al. Animal models of acute exacerbation of pulmonary fibrosis. Respir Res 24, 296 (2023). https://doi.org/10.1186/s12931-023-02595-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02595-z