
Abstract
Background
Trauma and a subsequent hemorrhagic shock (T/HS) result in insufficient oxygen delivery to tissues and multiple organ failure. Extracellular adenosine, which is a product of the extracellular degradation of adenosine 5’ triphosphate (ATP) by the membrane-embedded enzymes CD39 and CD73, is organ protective, as it participates in signaling pathways, which promote cell survival and suppress inflammation through adenosine receptors including the A2BR. The aim of this study was to evaluate the role of CD39 and CD73 delivering adenosine to A2BRs in regulating the host’s response to T/HS.
Methods
T/HS shock was induced by blood withdrawal from the femoral artery in wild-type, global knockout (CD39, CD73, A2BR) and conditional knockout (intestinal epithelial cell-specific deficient VillinCre-A2BRfl/fl) mice. At 3 three hours after resuscitation, blood and tissue samples were collected to analyze organ injury.
Results
T/HS upregulated the expression of CD39, CD73, and the A2BR in organs. ATP and adenosine levels increased after T/HS in bronchoalveolar lavage fluid. CD39, CD73, and A2BR mimics/agonists alleviated lung and liver injury. Antagonists or the CD39, CD73, and A2BR knockout (KO) exacerbated lung injury, inflammatory cytokines, and chemokines as well as macrophage and neutrophil infiltration and accumulation in the lung. Agonists reduced the levels of the liver enzymes aspartate transferase and alanine transaminase in the blood, whereas antagonist administration or CD39, CD73, and A2BR KO enhanced enzyme levels. In addition, intestinal epithelial cell-specific deficient VillinCre-A2BRfl/fl mice showed increased intestinal injury compared to their wild-type VillinCre controls.
Conclusion
In conclusion, the CD39-CD73-A2BR axis protects against T/HS-induced multiple organ failure.
Similar content being viewed by others


Introduction
Trauma resulting in hemorrhagic shock (T/HS) is a medical emergency that occurs when the body loses a considerable amount of blood. It leads to insufficient perfusion of essential organs and tissues due to the reduced blood volume [1,2,3]. Subsequent coagulopathy, endotheliopathy, microcirculatory dysfunction, inflammatory cell stimulation, and immunological activation result in multiple organ failure (MOF), where the injured organs include the lung, liver, and gut [4,5,6]. At the cellular level oxygen deficiency leads to the accumulation of lactic acid, inorganic phosphates, and free oxygen radicals [1].
ATP is a nucleotide found in both the extracellular and intracellular spaces where it is involved in important cellular biological processes. Cellular homeostasis is disrupted when intracellular ATP supplies decrease, which can also lead to cell death. ATP or ADP can be released from cells through membrane integrity loss, connexin/pannexin channels, and hormone-transporting vesicles [4, 7,8,9]. Adenosine is a nucleoside, which can be produced in the extracellular space through the degradation of ATP by ectonucleoside triphosphate diphosphohydrolase-1 (CD39), and ecto-5′-nucleotidase (CD73), which are membrane-associated enzymes. CD39 degrades extracellular ATP or ADP to AMP and CD73 degrades to AMP to adenosine. Also, adenosine can be directly released to the extracellular space via equibilirative nucleoside transporters (ENTs) [10,11,12,13,14,15]. Extracellular adenosine plays a role in regulating sleep, stroke, vasodilation, and inflammation [16]. Adenosine exerts its effects through binding to P1 or adenosine receptors (A1R, A2AR, A2BR, and A3R) and triggering intracellular signaling mechanisms such as up-or down-regulation of cyclic adenosine monophosphate (cAMP) [10, 16,17,18]. Adenosine receptors are found on many immune cell types such as neutrophils, monocytes, macrophages, dendritic cells, T cells, B cells, and NK cells, as well as on parenchymal cells, where they are involved in the regulation of inflammation [19, 20]. The A2BR is expressed on many immune and non-immune cell types, and in general, it has anti-inflammatory and tissue restorative functions [6, 20,21,22,23,24,25,26,27,28,29,30,31,32,33]. We have previously shown that the A2AR has a protective role against organ injury after T/HS [5, 6, 34]. However, the role of the A2BR and upstream CD39 and CD73 have not been explored. Here we investigated the role of the CD39-CD73-A2BR axis in regulating organ injury after T/HS using highly selective antagonists and agonists as well as global and conditional CD39, CD73, and A2BR deficient mice.
Materials and methods
Ethical statements and animals
All procedures on mice were performed under Columbia University Institutional Animal Care and Use Committee (IACUC) approval number (AABL4551/2021). Adult, male (8-12-week-old n = 4/group) CD39−/−, A2BR−/−, VillinCre-A2BARfl/fl, and their WT control VillinCre-A2BAR+/+ mice were bred at and wild-type C57BL/6J mice obtained from Charles River (Wilmington, MA, USA). CD73−/− (B6.129S1-Nt5etm1Lft/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). CD73−/− mice were bred and maintained in a specific pathogen-free Columbia University animal facility until being used in experiments. Mice had access to food and water ad libitum. They were kept in a room with a 12-h light-dark cycle under nonspecific pathogen-free conditions.
Drugs
The selective CD39 inhibitor sodium polyoxotungstate (POM1), CD73 inhibitor PSB 12,379 (N6-Benzyl-α,β-methyleneadenosine 5’-diphosphate disodium salt), and adenosine receptor agonist 1-(6-Amino-9 H-purin-9-yl)-1-deoxy-N-ethyl-β-D-ribofuranuronamide (NECA) were from Tocris (Bristol, UK). The CD39 mimic potato apyrase was from Sigma, and recombinant human (rh)CD73 was from Daresbury Proteins (Warrington, UK).
Study design and induction of trauma hemorrhagic shock
In one set of studies, wild-type mice were randomly assigned into the following groups: trauma/sham shock (T/SS) receiving vehicle (saline), trauma/hemorrhagic shock (T/HS) receiving vehicle (saline), T/HS receiving POM1 (5 mg/kg), T/HS receiving apyrase (125 U/kg), T/HS receiving PSB 12,379 (50 mg/kg), and T/HS receiving rhCD73 (2 mg/kg). In addition, in another group of studies, CD39−/−, CD73−/−, A2BAR −/− and wild-type type mice were subjected to T/SS or T/HS. Some CD39−/− and CD73−/− mice were pretreated with NECA (0.0025 mg/kg). In pharmacological experiments, mice were pretreated intraperitoneally (30 min before T/SS or T/HS) with various agents.
The mice were given anesthesia using 1% isoflurane and their rectal temperature was maintained between 36.5 and 37.5 °C using a feedback-controlled homeothermic blanket heating system (Sumno-suite, Kent Scientific). Hemorrhagic shock was induced using a fixed-pressure model [34] as described in the Additional data. Initially, a midline laparotomy of 2 cm was performed on the anesthetized mice, which was later closed with a 4 − 0 silk suture (034902, Covetrus, USA). Following this, catheters were placed in the right and left femoral arteries for monitoring blood pressure and blood withdrawal, respectively. A sterile 1-ml syringe with a 30G needle attached to PE-10 tubing filled with 0.2 ml of 1% heparinized saline was used for blood withdrawal, and each mouse received 1U heparin. Blood pressure was monitored using a continuous blood-pressure monitoring system (Powerlab 8/30, ADInstruments, Colorado Springs, CO, USA). After 5 min of baseline blood pressure recording, a drug or vehicle was administered to the mice, followed by inducing shock for a period of 2.5 h, during which the blood pressure was maintained between 28 and 32 mmHg by withdrawing or reinfusing the shed blood. At the end of the shock period, the mice were resuscitated with Ringer’s Lactate at three times the amount of shed blood for 15 min. Three hours after resuscitation, the mice were euthanized and bronchoalveolar lavage fluid (BALF), blood, and tissue samples were collected. T/SS animals were exposed to the same procedures as other animals except for blood withdrawal (Additional file 1: Fig. S1).
Tissue preparation for evaluating neutrophil sequestration, mouse cytokine array, and western blot analysis
Lung, liver, kidney, and gut samples were pooled from 4 mice in the same group, homogenized, sonicated, and treated with a 1X RIPA Buffer with Protease/phosphatase inhibitor cocktail (P8340, Sigma, USA), and the resulting homogenate was centrifuged at 13.000 x g for 10 min at 4 °C. Total protein content was determined by using Qubit 4.0 Fluorometer (Thermo Fisher, USA) as per the manufacturer’s instructions.
Collection of BALF samples and determination of BALF ATP, Adenosine, and cAMP levels
Three hours after the end of resuscitation the mice were euthanized, and the trachea was isolated for collecting BALF samples. Briefly, after a small incision a syringe with a 23G needle filled with 1 ml of sterile saline was inserted into the trachea. After centrifugation (1500 x g for 15 min at 4 °C) the supernatant was used for the assays [34]. ATP (ab83355, Abcam, USA) and cAMP (KGE012B, R&D Systems, USA) in BALF were measured by a colorimetric assay. Adenosine was measured by a fluorometric assay (ab211094, Abcam, USA) according to the manufacturer’s instructions.
Lung permeability measurements
Mice were re-anesthetized with isoflurane 3 h after the end of resuscitation. Evans blue dye (EBD) technique was used to determine lung permeability. EBD was administered through the tail vein, and about 1 ml of blood was withdrawn from the tail artery five minutes later. The supernatant of BALF was assayed at 620 nm spectrophotometrically. The amount of Evans blue dye in the BALF was then expressed as a proportion of the amount in the plasma [5, 34].
Determination of pulmonary neutrophil sequestration
To evaluate neutrophil sequestration in the lung following T/SS or T/HS, myeloperoxidase (MPO) activity was assessed using an MPO activity kit (MAK068, Sigma, USA). The supernatant of lung lysates was assayed according to the manufacturer’s instructions [34].
Mouse cytokine array
To examine cytokine and chemokine expression in lung samples, a total of 200 µg protein was analyzed using the Proteome Profiler Mouse Cytokine Array Panel A Kit (ARY006, R&D Systems, USA). The expression of cytokines and chemokines was determined densitometrically using Fiji software after subtracting the negative control’s average signal. Values were expressed as % of T/SS [35].
Immunofluorescence staining
Macrophage staining was performed as previously described [35]. Lung section (5 μm) were fixed in 4% paraformaldehyde in PBS, washed, and immersed for 30 min in 0.1 M PBS containing 0.3% Triton-X-100 (PBS-T)/10% normal goat serum. Sequentially 2 lung sections from each sample were incubated overnight at 4 °C with AlexaFlour 488-conjugated monoclonal mouse anti-F4/80 (53-4801-82, Thermo Fisher, USA). The following day, sections were incubated with 4′,6-diamidino-2-phenylindole (DAPI) (D9542, Sigma, USA) at room temperature for 5 min. Sections were analyzed using confocal laser scanning microscopy (Zeiss LSM 900, Jena, Germany). Eight different regions of interest (ROI) from the sections were calculated. The percentage of macrophages in the lung tissue was calculated by dividing the results obtained from T/HS by the results of T/SS and then multiplying the quotient by 100.
Western blot
CD39, CD73, A2BR, phosphorylated-Phosphatase and Tensin (p-PTEN) and matrix metalloproteinase-9 (MMP-9) protein levels after T/SS or T/HS were determined using western blotting of pooled samples run three times. Thirty micrograms of protein per sample were size-fractioned using 4–20% Mini-Protean TGX Stain-Free (4,568,093, Bio-Rad, Life Sciences Research) electrophoresis gel and then transferred to a PVDF membrane (1,620,174, Bio-Rad, Life Sciences Research) using Mini Trans-Blot Electrophoretic Transfer System (1,703,930, Bio-Rad, Life Sciences Research). The membranes were first blocked with a blocking solution composed of 5% non-fat dry milk in 50 mM Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature. Following this, the membranes were washed with 50 mM TBS-T and then incubated overnight with a rabbit monoclonal anti-CD39 (ab223842, Abcam, USA), rabbit polyclonal anti-CD73 (ab175396, Abcam, USA), rabbit polyclonal anti-A2BR (ab229671, Abcam, USA), rabbit polyclonal p-PTEN (9551, Cell Signaling, USA) and rabbit polyclonal anti-MMP-9 (ab38898) at a dilution 1:1000, 1:1000, 1:500, 1:1000 and 1:2000 respectively. The following day, the membranes were washed with TBS-T and then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (ab97051, Abcam, USA) at a dilution of 1:5000 in the blocking solution for 2 h at room temperature. To ensure the protein loading was consistent, the membranes were stripped and re-probed with an HRP-conjugated anti-ß actin antibody (ab20272, Abcam, USA). The membranes were then developed using Clarity Western ECL Substrate Kit (1,708,280, Bio-Rad, Life Sciences Research) [34].
Measurement of AST and ALT levels
Plasma samples were analyzed to determine the levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). To do this, the samples were diluted with AST (TR70121 Thermo Fisher, USA) and ALT (TR71122 Thermo Fisher, USA) reagents at a 1:10 ratio, and the resulting light signal was measured using a spectrophotometer at 340 and 405 nm [34].
Histopathological assessment of lung and intestinal injury
Sections of lung and gut were cut at 5 μm thickness, stained with the hematoxylin-eosin (H&E) method, and scanned with a Lecia AT2 slide scanner (Leica, USA). Lung sections were evaluated histopathologically in terms of the parameters: 1: neutrophils in the alveolar spaces, 2: neutrophils in the interstitial space, 3: hyaline membranes, 4: proteinaceous debris filling in the airspaces, 5: alveolar septal thickening by a blind observer [36]. Ileum samples were evaluated histopathologically in terms of the parameters: 1: desquamation and necrosis of upper 1/3 villi, 2: progressive peel off mid of villi, 3: peel of lower 1/3 villi and necrosis of cript cells, 4: necrosis of 2/3 cript cells, 5: complete loss of basal cripts.
Determination of IL-10 and IL-6 levels by enzyme-linked immunosorbent assay
IL-6 and IL-10 levels were measured in plasma samples using enzyme-linked immunosorbent assay (ELISA) kits (DY406, DY417; R&D Systems Minneapolis, USA) according to the manufacturer’s instructions [34].
Statistical analysis
The Shapiro-Wilk test was used to determine the normality of the data in each group, which demonstrated a normal distribution. Statistical analysis was conducted using Graph-Pad Prism (San Diego, CA, USA). One-way ANOVA or t-test was performed followed by Tukey’s test as appropriate. The results were presented as mean ± S.D. values, and a p-value of less than 0.05 was considered statistically significant.
Results
T/HS increases CD39, CD73, and A2BR expression in lung, liver, kidney, and gut
T/HS increased the expression of CD39, CD73, and the A2BR in the lung, liver, kidney, and gut when compared to T/SS (Fig. 1, A-C). Of all the organs investigated, CD39, CD73 and the A2BR expression was highest in the lung. Thus in subsequent experiments, our main but not exclusive focus was on lung injury.

T/HS increases CD39, CD73, and A2BAR expression in lung, liver, kidney, and gut. A–C CD39, CD73, and A2BAR expression were evaluated using western blotting of protein extracts. Data are mean ± S.D. (n = 4/group). **p < 0.01 compared with T/SS
Role of CD39, CD73, and A2BR in regulating ATP, adenosine, and cAMP levels after T/HS
We found that T/HS increased ATP, adenosine, and cAMP levels in BALF samples (Fig. 2A–C). CD39, CD73, and A2BR KO further increased T/HS-induced ATP and adenosine (Fig. 2A, B). T/HS increased cAMP compared to T/SS and cAMP levels decreased in CD39, CD73, and A2BR KO mice in T/HS BALF (Fig. 2C).

ATP (A), adenosine (B), and cAMP (C) levels in BALF following T/HS in the CD39, CD73, and A2BAR KO samples. ATP and cAMP were measured colorimetrically and adenosine was measured by a fluorometric assay. Data are mean ± S.D. (n = 4/group) *p < 0.05 compared with T/SS-V, **p < 0.01 compared with T/SS-V, #p < 0.05 compared with T/HS-V, ##p < 0.01 compared with T/HS-V
CD39, CD73, and the A2BR suppress lung permeability and neutrophil infiltration
Treatment with the CD39 antagonist POM1 exacerbated and treatment with the CD39-like enzyme apyrase moderated T/HS-induced lung injury and neutrophil sequestration after T/HS (Fig. 3A, B). KO of CD39, CD73, and the A2BR increased lung permeability and neutrophil sequestration (Fig. 3C–H). The CD73 antagonist PSB 12,379 increased lung injury and neutrophil infiltration while rhCD73 decreased these parameters (Fig. 3E, F). We then examined whether adenosine receptor stimulation was able to rescue the increased injury in CD39−/− and CD73−/− mice. NECA pretreatment decreased neutrophil infiltration in CD39−/− and CD73−/− mice after T/HS (Fig. 4A, B). The lung injury score increased in T/HS vs. T/SS mice and was further increased in the CD39, CD73, and A2BR KO mice (Fig. 5A, B).

CD39, CD73, and A2BR regulation of lung permeability and MPO activity. Lung permeability was determined using the EBD method (A, C, E, G) and MPO activity as a surrogate for neutrophil sequestration (B, D, F, H) was determined spectrophotometrically. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with corresponding T/SS, **p < 0.01 compared with corresponding T/SS, #p < 0.05 compared with corresponding T/HS-V, ##p < 0.01 compared with corresponding T/HS

Effect of NECA on lung MPO activity in CD39−/− and CD73−/− mice. MPO activity was determined from the lung spectrophotometrically (A, B). Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS

Lung injury was induced by T/HS. Representative images of H&E-stained lung samples (A). Pulmonary injury was evaluated using 5 independent parameters (Neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris filling the airspaces and alveolar septal thickness) (B). Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with T/HS-V, ##p < 0.01 compared with T/HS-V
Regulation of lung cytokine levels by CD39, CD73, and the A2BR
Using an antibody array, we assessed the relative expression levels of 40 different cytokines and chemokines including CXCL13/BLC/BCA-1, C5a, G-CSF, GM-CSF, CCL1/I-309, CCL11/Eotaxin, ICAM-1, IFN-gamma, IL-1 alpha/IL-1F1, IL-1 beta/IL-1F2, IL-1ra/IL-1F3, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12 p70, IL-13, IL-16, IL-17, IL-23, IL-27, CXCL10/IP-10, CXCL11/I-TAC, CXCL1/KC, M-CSF, CCL2/JE/MCP-1, CCL12/MCP-5, CXCL9/MIG, CCL3/MIP-1 alpha, CCL4/MIP-1 beta, CXCL2/MIP-2, CCL5/RANTES, CXCL12/SDF-1, CCL17/TARC, TIMP-1, TNF-alpha, and TREM-1 in the lung (Fig. 6A). We found that levels of pro-inflammatory cytokines and chemokines (BLC, C5, G-CSF, SICAM-1, IL-1α, IL-1ß, IL-1ra, IL-6, IL-16, IP-10, KC, M-CSF, MCP-1, MCP-5, MIG, MIP-1α, MIP-1ß, MIP-2, RANTES, TNF- α and TREM-1) increased in the CD39, CD73, and A2BRs KO mice in the lung after T/HS (Fig. 6B, C).

Quantification of pro- and anti-inflammatory cytokines and chemokines in the lung. Representative images of cytokine-chemokine arrays that were used to interrogate cytokine expression (A). Proteome Profiler Mouse Cytokine-Chemokine array was used to analyze cytokines in mice subjected to T/HS. Data were visualized by transforming them into a heat map B and also expressed as a bar graph (C). Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with T/HS-V, ##p < 0.01 compared with T/HS-V
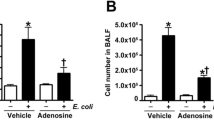
CD39, CD73, and A2BR moderate macrophage accumulation in the lung
CD39, CD73, and A2BR deficiency all increased macrophage accumulation following T/HS, as indicated by immunofluorescence (Fig. 7).

Macrophage accumulation in the lung after T/HS. Accumulated macrophages were determined by F4/80 immunostaining in the lung and representative images are shown as well as averages and means of macrophage counts in the various groups. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with corresponding T/HS-V, ##p < 0.01 compared with corresponding T/HS
Effect of CD39, CD73 and A2BR KO on the expression of survival-related proteins
To investigate the role of CD39, CD73, and the A2BR in regulating the expression of survival-related proteins, we analyzed p-PTEN and MMP-9 expression after T/HS. CD39, CD73, and A2BRs KO all decreased p-PTEN (Fig. 8A) and increased MMP-9 (Fig. 8B) expression levels in lung tissue.

Expression levels of survival-related kinases after T/HS. p-PTEN A and MMP-9 B expression levels were determined using western blot. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with corresponding T/HS-V, ##p < 0.01 compared with corresponding T/HS
Regulation of intestinal injury by CD39, CD73, and the A2BR
T/HS increased gut injury compared to T/SS (Additional file 2: Fig. S2). Global CD39, CD73, and A2BR deficiency failed to influence the extent of gut injury in T/HS mice (Fig. 2). However, VillinCre-A2BARfl/fl mice had increased gut injury when compared to their WT control VillinCre-A2BAR+/+ mice after T/HS. (Additional file 3: Fig. S3).
Role of CD39, CD73 and A2BR in regulating T/HS-induced liver injury
Apyrase or rhCD73 treatment suppressed T/HS-enhanced AST and ALT levels, indicating decreased liver injury, while POM1 and PSB 12,379 increased AST and ALT (Additional file 4: Fig. S4A, B). In addition, KO of CD39, CD73, and A2BRs led to elevated AST and ALT levels (Additional file 4: Fig. S4C, D).
Role of CD39, CD73 and A2BRs in regulating plasma IL-6 and IL-10 levels
As compared to vehicle-treated T/HS mice, POM1-treated mice had higher plasma IL-6 levels and similar plasma IL-10 levels. Also, apyrase-treated T/HS mice had lower plasma IL-6 levels and increased plasma IL-10 levels compared to vehicle-treated T/HS mice (Additional file 5: Fig. S5A, B). CD39−/− mice after T/HS had increased IL-6 and decreased IL-10 compared to wild-type controls (Additional file 5: Fig. S5C, D).
When compared to vehicle-treated T/HS mice, PSB 12,379-treated T/HS animals had increased plasma IL-6 and reduced IL-10 levels and rhCD73-treated T/HS mice had decreased plasma IL-6 and increased plasma IL-10 (Figures E6E and E6F). Plasma IL-6 was increased and IL-10 decreased in CD73−/− compared to wild-type T/HS mice (Additional file 5: Fig. S5E, F). A2BAR−/− mice exhibited higher plasma IL-6 levels while having lower plasma IL-10 levels compared to the A2BR+/+ group subjected to T/HS (Additional file 5: Fig. S5G, H).
Discussion
We recently demonstrated that exogenous stimulation of the A2AR decreases organ injury after T/HS. In addition, we found that inhibition or KO of the A2AR leads to increased lung permeability, MPO level, and augmented liver enzymes [34]. Our current studies demonstrate for the first time that ATP is released into the extracellular space during T/HS. In agreement with previous studies implicating extracellular ATP as the primary source for adenosine generation in ischemia of particular organs [4, 7,8,9], our studies targeting CD39 and CD73 demonstrate that ATP is also the likely source of extracellular adenosine following T/HS, a condition whose pathophysiology is much more complex than that of ischemia of the various organs. In addition, we demonstrate for the first time that the A2BR activated by endogenous adenosine protects organs from T/HS-induced organ injury.
CD39 is a transmembrane protein found in the spleen, thymus, lung, and placenta and is largely linked with immune cell populations and endothelial cells [10, 37, 38]. Several proinflammatory cytokines, oxidative stress, and hypoxia affect the CD39 expression [39]. Studies indicate that CD39 is organ protective against ischemia/reperfusion injury, sepsis, and heart disease [40,41,42,43,44,45,46,47]. For example, in CD39 deficient mice the organ injury and inflammation that followed cardiac [44], renal [41], hepatic [18, 42], and intestinal [48] ischemia-reperfusion injury were more severe than in the corresponding wild-type mice. A common thread in these studies was that exogenous supplementation with potato apyrase reversed the increased ischemic organ injury of CD39 deficient mice. Potato apyrase is widely used as a CD39 mimic, because it has an amino acid sequence that is highly homologous to CD39, particularly within its four apyrase-conserved regions [49]. Given that T/HS causes global ischemia-reperfusion injury, our results demonstrating protective effects for CD39 in T/HS correspond well with earlier results with ischemia-reperfusion injury of the various organs. Given the commonalities of mechanisms for the injury of the various organs, it will be of interest to determine whether protection in the various models can be traced back to one particular cell type expressing CD39 or whether protection depends on different cell types based on the organ.
CD73 regulates multiple events such as cellular hemostasis and tissue injury and is found in a variety of tissues, including the colon, brain, kidney, liver, lung, and heart [50]; on leukocytes derived from peripheral blood, spleen, lymph nodes, thymus, and bone marrow [50]; as well as on endothelium [51]. The expression and function of this enzyme are upregulated under hypoxic conditions [52, 53], and by several proinflammatory mediators, such as transforming growth factor (TGF)-β, interferons (IFNs), TNF-α, IL-1β, and prostaglandin E2 [54, 55]. Similar to CD39, CD73 is also organ protective against hypoxic and ischemic conditions [50, 53, 56]. The increased vascular permeability and polymorphonuclear neutrophil extravasation noted in hypoxic CD73 deficient mice were reversed by stimulating ARs by exogenous administration of 5′-(N-ethylcarboxamido) adenosine (NECA), a general AR agonist, or by exogenous reconstitution with a soluble CD73-like nucleotidase [50]. In this respect, our data with the NECA-mediated “rescue” of the increased neutrophil infiltration of lungs observed in CD73 (and CD39) deficient mice indicate that CD73 together with CD39 deliver adenosine to adenosine receptors to dampen inflammation and injury in T/HS.
The A2BR activates intracellular signaling mechanisms such as cAMP [57]. Elevated intracellular levels of cAMP are frequently linked to anti-inflammatory outcomes, including the synthesis of IL-10, the inhibition of leukocyte infiltration, and pro-inflammatory cytokines [58]. We have previously shown that A2BR-deficient mice have higher mortality and increased pro-inflammatory cytokines such as IL-6, TNF-α and MIP-2 in sepsis [59]. Also, posttreatment with the A2BR agonist BAY 60-6583 decreases lung permeability but not neutrophil infiltration into the lung in a rat model of T/HS [6]. The current studies confirm the generally protective role of A2BR in a global ischemia-reperfusion model.
There are several outstanding questions that require further studies to answer. In our view, the most pressing of these are (1) what organs and cells release ATP in T/HS, (2) what are the release mechanisms, and (3) which cell types expressing CD39, CD73, and A2BR mediate protection.
In conclusion, our work indicates the organ-protective effects of the purinergic system in a T/HS model. The results point to the purinergic system as a therapeutic target to treat traumatic hemorrhagic shock.
Availability of data and materials
Not applicable.
References
Cannon JW, Hemorrhagic Shock. N Engl J Med. 2018;19:1852–3.
Huan Z, Tang Y, Xu C, Cai J, Yao H, Wang Y, Bu F, Ge X. PTPRO knockdown protects against inflammation in hemorrhage shock-induced lung injury involving the NF-kappaB signaling pathway. Respir Res. 2022;1:195.
Kao RL, Xu X, Xenocostas A, Parry N, Mele T, Martin CM, Rui T. Induction of acute lung inflammation in mice with hemorrhagic shock and resuscitation: role of HMGB1. J Inflamm (Lond). 2014;1:30.
Valade G, Libert N, Martinaud C, Vicaut E, Banzet S, Peltzer J. Therapeutic potential of mesenchymal stromal cell-derived extracellular vesicles in the Prevention of Organ Injuries Induced by Traumatic hemorrhagic shock. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.749659.
Hasko G, Xu DZ, Lu Q, Nemeth ZH, Jabush J, Berezina TL, Zaets SB, Csoka B, Deitch EA. Adenosine A2A receptor activation reduces lung injury in trauma/hemorrhagic shock. Crit Care Med. 2006;4:1119–25.
Koscso B, Trepakov A, Csoka B, Nemeth ZH, Pacher P, Eltzschig HK, Hasko G. Stimulation of A2B adenosine receptors protects against trauma-hemorrhagic shock-induced lung injury. Purinergic Signal. 2013;3:427–32.
Chen Y, Bao Y, Zhang J, Woehrle T, Sumi Y, Ledderose S, Li X, Ledderose C, Junger WG. Inhibition of neutrophils by Hypertonic saline involves Pannexin-1, CD39, CD73, and other Ectonucleotidases. Shock. 2015;3:221–7.
Ham PB, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol. 2017. https://doi.org/10.1016/j.pneurobio.2016.06.006.
Wang Y, Copeland J, Shin M, Chang Y, Venton BJ. CD73 or CD39 deletion reveals different mechanisms of formation for spontaneous and mechanically stimulated Adenosine and Sex Specific Compensations in ATP degradation. ACS Chem Neurosci. 2020;6:919–28.
Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;6:355–67.
Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic signaling, DAMPs, and inflammation. Am J Physiol Cell Physiol. 2020;5:C832–5.
Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;9:759–70.
Zhang X, Du P, Luo K, Li Y, Liu Z, Wang W, Zeng C, Ye Q, **ao Q. Hypoxia-inducible factor-1alpha protects the liver against ischemia-reperfusion injury by regulating the A2B adenosine receptor. Bioengineered. 2021;1:3737–52.
Tiwari-Heckler S, Lee GR, Harbison J, Ledderose C, Csizmadia E, Melton D, Zhang Q, Junger W, Chen G, Hauser CJ, Otterbein LE. Longhi, and SC Robson. Extracellular mitochondria drive CD8 T cell dysfunction in trauma by upregulating CD39. Thorax. 2023;2:151–9.
Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;24:2322–33.
Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets—what are the challenges? Nat Rev Drug Discovery. 2013;4:265–86.
Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. Faseb J. 2009;2:473–82.
Wang S, Gao S, Zhou D, Qian X, Luan J, Lv X. The role of the CD39-CD73-adenosine pathway in liver disease. J Cell Physiol. 2021;2:851–62.
Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;3:201–12.
Rai R. Cross talk of purinergic and immune signaling: implication in inflammatory and pathogenic diseases, in purinergic system, Bagatini MD, Ed., 2022, IntechOpen: United Kingdom. 2022; p. 1–29.
Sun Y, Huang PB. Adenosine A(2B) receptor: from cell biology to human diseases. Front Chem. 2016. https://doi.org/10.3389/fchem.2016.00037.
Xu XT, Zhu QW, Niu FF, Zhang R, Wang Y, Wang WY, Sun DW, Wang XT, Wang AZ. A2BAR activation attenuates acute lung injury by inhibiting alveolar epithelial cell apoptosis both in vivo and in vitro. Am J Physiol-Cell Ph. 2018;4:C558–70.
Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, Kepka-Lenhart D, Morris SM Jr, Gause WC, Leibovich SJ, Hasko G. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012;1:376–86.
Koscso B, Csoka B, Selmeczy Z, Himer L, Pacher P, Virag L, Hasko G. Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J Immunol. 2012;1:445–53.
Csoka B, Koscso B, Toro G, Kokai E, Virag L, Nemeth ZH, Pacher P, Bai P, Hasko G. A2B Adenosine Receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes. 2014;3:850–66.
Nemeth ZH, Lutz CS, Csoka B, Deitch EA, Leibovich SJ, Gause WC, Tone M, Pacher P, Vizi ES, Hasko G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;12:8260–70.
Patel N, Wu W, Mishra PK, Chen F, Millman A, Csoka B, Koscso B, Eltzschig HK, Hasko G, Gause WC. A2B adenosine receptor induces protective antihelminth type 2 Immune responses. Cell Host Microbe. 2014;3:339–50.
Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A. Szabo. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000;13:2065–74.
Csoka B, Nemeth ZH, Virag L, Gergely P, Leibovich SJ, Pacher P, Sun CX, Blackburn MR, Vizi ES, Deitch EA, Hasko G. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;7:2685–95.
Csoka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, Ledent C, Deitch EA, Spolarics Z, Nemeth ZH, Hasko G. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J. 2008;10:3491–9.
Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;10:4634–40.
Hasko G, Nemeth ZH, Vizi ES, Salzman AL, Szabo C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol. 1998;3:261–8.
Szabo C, Scott GS, Virag L, Egnaczyk G, Salzman AL, Shanley TP, Hasko G. Suppression of macrophage inflammatory protein (MIP)-1alpha production and collagen-induced arthritis by adenosine receptor agonists. Br J Pharmacol. 1998;2:379–87.
Kelestemur T, Nemeth ZH, Pacher P, Antonioli L, Hasko G. A 2a Adenosine Receptors regulate multiple organ failure after hemorrhagic shock in mice. Shock. 2022;4:321–31.
Beker MC, Caglayan AB, Altunay S, Ozbay E, Ates N, Kelestemur T, Caglayan B, Kilic U, Doeppner TR, Hermann DM, Kilic E. Phosphodiesterase 10A is a critical target for Neuroprotection in a mouse model of ischemic stroke. Mol Neurobiol. 2022;1:574–89.
Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. Acute Lung Injury in Animals Study. An official american thoracic society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;5:725–38.
Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JS, Imai M, Edelberg JM, Rayburn H, Lech M, Beeler DL, Csizmadia E, Wagner DD, Robson SC, Rosenberg RD. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med. 1999;9:1010–7.
Mizumoto N, Kumamoto T, Robson SC, Sevigny J, Matsue H, Enjyoji K, Takashima A. CD39 is the dominant Langerhans cell associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat Med. 2002;4:358–65.
Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;1:224–32.
Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;13:3986–92.
Grenz A, Zhang H, Hermes M, Eckle T, Klingel K, Huang DY, Muller CE, Robson SC. Osswald, and HK Eltzschig. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J. 2007;11:2863–73.
Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol. 2010;7:4017–24.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;2:138–50.
Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Muller CE, Eltzschig HK. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;16:1784–94.
Leite RO, Pods CB, Haas F, da Silveira KR, Mohr APS, Bertoni MS, Soares JH, Azambuja M, Dal Prá LLP, da Cruz NE, Gelsleichter K, Begnini G, Hasko MR, Wink RM, Spanevello E. Braganhol ATPergic signaling disruption in human sepsis as a potential source of biomarkers for clinical use. Clin Exp Med. 2023. https://doi.org/10.1007/s10238-023-01045-w.
Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;4:460–7.
Sayegh MN, Cooney KA, Han WM, Cicka M, Strobel F, Wang L, Garcia AJ, Levit RD. Hydrogel delivery of purinergic enzymes improves cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2023. https://doi.org/10.1016/j.yjmcc.2023.02.001.
Hart ML, Henn M, Kohler D, Kloor D, Mittelbronn M, Gorzolla IC, Stahl GL, Eltzschig HK. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008;8:2784–97.
Handa M, Guidotti G. Purification and cloning of a soluble ATP-diphosphohydrolase (apyrase) from potato tubers (Solanum tuberosum). Biochem Biophys Res Commun. 1996;3:916–23.
Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5’-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;11:1395–405.
Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5’-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998;8:1433–43.
Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;5:783–96.
Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;7:993–1002.
Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;5:231–7.
Regateiro FS, Howie D, Nolan KF, Agorogiannis EI, Greaves DR, Cobbold SP, Waldmann H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-beta. Eur J Immunol. 2011;10:2955–65.
Hasko G, Csoka B, Koscso B, Chandra R, Pacher P, Thompson LF, Deitch EA, Spolarics Z, Virag L, Gergely P, Rolandelli RH, Nemeth ZH. Ecto-5’-nucleotidase (CD73) decreases mortality and organ injury in sepsis. J Immunol. 2011;8:4256–67.
Zhao N, **a G, Cai J, Li Z, Lv XW. Adenosine receptor A2B mediates alcoholic hepatitis by regulating cAMP levels and the NF-KB pathway. Toxicol Lett. 2022. https://doi.org/10.1016/j.toxlet.2022.01.012.
Tavares LP, Negreiros-Lima GL, Lima KM, Pinho ES, Teixeira MM, Sousa LP. Blame the signaling: role of cAMP for the resolution of inflammation. Pharmacol Res. 2020. https://doi.org/10.1016/j.phrs.2020.105030.
Csoka B, Nemeth ZH, Rosenberger P, Eltzschig HK, Spolarics Z, Pacher P, Selmeczy Z, Koscso B, Himer L, Vizi ES, Blackburn MR, Deitch EA, Hasko G. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol. 2010;1:542–50.
Funding
This study was funded by NIH (R01HL158519-GH, R01GM066189-GH, R01DK113790-GH).
Author information
Authors and Affiliations
Contributions
All authors contributed to the completion of this study. TK, and GH designed the study. TK carried out animal studies, experimental work, analyzed data and wrote the manuscript. JB provided recombinant human protein. SCR provided floxed mice. TK, ZHN, PP, EKH, and GH outlined the study subject and critically edited the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
All procedures on mice were performed under Columbia University Institutional Animal Care and Use Committee (IACUC) approval number (AABL4551/2021).
Consent for publication
Not applicable.
Competing interests
G.H. owns stock in Purine Pharmaceuticals, Inc. S.C.R. is the scientific cofounder of Purinomia and a consultant to SynLogic and eGenesis. The other authors have no financial conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Experimental design. Mice are exposed to T/SS or T/HS for 2.5 h. Mice received vehicle, agonist, or antagonist 30 min before shock induction. After a 15-min resuscitation period and a subsequent observation period of 3 hours, mice were euthanized and tissues were collected
Additional file 2: Fig. S2.
Assessment of intestinal injury after T/HS. Effects of CD39, CD73 and A2BR deficiency are shown. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS.
Additional file 3. Fig. S3.
Determination of intestinal injury after T/HS in IEC-specific A2BR deficient mice. Results with IEC-specific VillinCre-A2BRfl/fl mice and control are shown. Data are mean ± S.D. (n = 4/group). **p < 0.05 compared with T/HS-VillinCre-A2BAR+/+.
Additional file 4. Fig. S4.
CD39, CD73, and A2BR regulation of liver enzymes. Aspartate aminotransferase (AST) (A, C) and alanine aminotransferase (ALT) (B, D) levels were determined from plasma spectrophotometrically. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with corresponding T/HS-V, ##p < 0.01 compared with corresponding T/HS.
Additional file 5. Fig. S5.
Quantification of cytokine levels in plasma. IL-6 (A, C, E, G) and IL-10 (B, D, F, H) in blood 3 h following resuscitation were determined using ELISA. Data are mean ± S.D. (n = 4/group). *p < 0.05 compared with T/HS, **p < 0.05 compared with T/HS, #p < 0.05 compared with corresponding T/HS-V, ##p < 0.01 compared with corresponding T/HS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kelestemur, T., Németh, Z.H., Pacher, P. et al. Adenosine metabolized from extracellular ATP ameliorates organ injury by triggering A2BR signaling. Respir Res 24, 186 (2023). https://doi.org/10.1186/s12931-023-02486-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02486-3