
Abstract
Background
Laryngeal cancer (LC) is a malignant tumor with high incidence and mortality. We aim to explore key genes as novel biomarkers to find potential target of LC in clinic diagnosis and treatment.
Methods
We retrieved GSE143224 and GSE84957 datasets from the Gene Expression Omnibus database to screen the differentially expressed genes (DEGs). Hub genes were identified from protein-protein interaction networks and further determined using receiver operating characteristic curves and principal component analysis. The expression of hub gene was verified by quantitative real time polymerase chain reaction. The transfection efficiency of BCL2 interacting protein like (BNIPL) was measured by western blot. Proliferation, migration, and invasion abilities were detected by Cell Counting Kit-8, wound-healing, and transwell assays, respectively.
Results
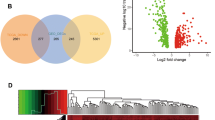
Total 96 overlap** DEGs were screened out from GSE143224 and GSE84957 datasets. Six hub genes (BNIPL, KRT4, IGFBP3, MMP10, MMP3, and TGFBI) were identified from PPI network. BNIPL was selected as the target gene. The receiver operating characteristic curves of BNIPL suggested that the false positive rate was 18.5% and the true positive rate was 81.5%, showing high predictive values for LC. The expression level of BNIPL was downregulated in TU212 and TU686 cells. Additionally, overexpression of BNIPL suppressed the proliferation, migration, and invasion of TU212 and TU686 cells.
Conclusion
BNIPL is a novel gene signature involved in LC progression, which exerts an inhibitory effect on LC development. These findings provide a novel insight into the pathogenesis of LC.
Similar content being viewed by others


Introduction
Laryngeal cancer (LC) is a kind of malignancy of head and neck with high incidence, with approximately 1.7 million cases of LC and nearly 90,000 deaths worldwide each year [1]. More than 40% of LC patients with LC are diagnosed at an advanced stage, often with spread to the supraglottic and para glottic space [2, 3]. The 5-year overall survival rate for LC shows continued decline in the last few decades [4]. The occurrence of LC is multifactorial and is related to common carcinogens (tobacco and alcohol) [5]. Radiotherapy, chemotherapy, and surgical resection are the main treatments for LC, but the recurrence rate of advanced LC is 25-50%, and there are still numerous patients with poor prognosis [6, 7]. Therefore, screening the promising biomarkers and therapeutic targets for the early diagnosis of LC has become a hot topic.
Currently, the potential for genomic data sharing plays a crucial role in cancer research [8]. The identification of genes involved in cancer progression holds great promise for medical diagnosis [9]. After retrieving quantitative and clinical gene expression data of LC from the Cancer Genome Atlas and Gene Expression Omnibus (GEO) database, the role of B7-H3 in the diagnosis and prognosis of LC is determined [10]. Based on the robust rank aggregation analysis for the consolidation of gene expression datasets from the GEO database for patients with LC, CDK1, PC24, HOXB7, and SELENBP1 are screened as potential biomarkers of LC [11]. Therefore, searching for potential gene markers as the therapeutic targets for LC is utmost essential.
BCL2 interacting protein like (BNIPL) is a pro-apoptotic gene that can maintain homeostasis between cell proliferation and apoptosis in vivo [12]. There are 2 spliceosomes of BNIPL, BNIPL-1 and BNIPL-2. Overexpression of BNIPL-2 can significantly inhibit tumor growth by regulating molecules related to cell proliferation and apoptosis of hepatocellular carcinoma cells [Full size image
Functional enrichment analysis
The most significantly enriched top 6 GO terms were screened. The DEGs were significantly enriched in “extracellular matrix organization”, “keratinization”, “proteolysis”, “collagen catabolic process”, “epidermis development”, “extracellular matrix disassembly” in BP; “extracellular region”, “extracellular space”, “extracellular exosome”, “extracellular matrix”, “cornified envelope”, “apical pl1asma membrane” in CC; “serine-type endopeptidase activity”, “CXCR3 chemokine receptor binding”, “metalloendopeptidase activity”, “chemokine activity, protease binding”, and “cytokine activity” in MF. The top 6 GO enrichment results were displayed in bar chart and bubble chart (Fig. 3A, C).

Enrichment analysis of common DEGs. (A) Gene Ontology (GO) enrichment bar chart. The abscissa is GO term and the ordinate is -log10 (p-value) of enrichment in each term. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) bar chart; (C) GO enrichment analysis bubble map, color depth of each node represents corrected p value, the size of the node refers to the number of genes involved; (D) KEGG bubble diagram
KEGG pathway analysis suggested that the enriched pathways mainly included “IL-17 signaling pathway”, “Cytokine-cytokine receptor interaction”, “Viral protein interaction with cytokine and cytokine receptor”, “Toll-like receptor signaling pathway”, “Ether lipid metabolism”, “Chemokine signaling pathway”, “ECM-receptor interaction”. The most significantly enriched KEGG pathways were displayed in bar chart and bubble chart (Fig. 3B, D).
PPI network construction and identification of hub genes
The PPI networks of DEGs were visualized using Cytoscape (Fig. 4A). We identified 6 hub genes (BNIPL, KRT4, IGFBP3, MMP10, MMP3, and TGFBI) from the key module of PPI networks (Fig. 4B).

Protein-protein interaction network and the identification of hub genes. (A) The protein-protein interaction network of DEGs. (B) The key module where BNIPL resides. The lines between the nodes represent the interactions between genes
The GO enrichment results that the hub genes enriched in were mainly “extracellular matrix” and “negative regulation of cell proliferation” (Fig. 5A). PCA analysis showed that the variance interpretation rate of PC1 and PC2 was 87.2%, indicating that hub genes could distinguish between the LC samples from control samples. The scatter plot suggested that the two groups had a good separation, further confirming the validity of the two principal components (Fig. 5B). The expression levels of hub genes in the original sample of GSE84957 dataset were shown in the ridgeline plot (Fig. 5C).

Analysis of hub genes. (A) GO enrichment chord map, including three parts: logFC is fold-change of genes; other columns are the GO term, and the different links of the gene indicate whether the gene is in this GO term. (B) Principal component analysis of hub genes. PC1 and PC2 on the axes in the figure are the first and second principal components (the variance explanation rate of differences by potential variables); Dots represent samples, and different colors represent different groups. (C) Expression of ridgeline plot of hub gene. The horizontal coordinate is the gene expression, the shape of the mountain represents the dispersion between a set of data, and the height is the number of samples corresponding to the gene expression
Analysis of hub genes
We plotted the ROC curves of BNIPL, KRT4, IGFBP3, MMP10, MMP3, and TGFBI using the raw data of GSE84957 dataset (Fig. 6). The false positive rates of ROC curves of BNIPL, KRT4, IGFBP3, MMP10, MMP3 and TGFB were 18.5%, 8.6%, 1.2%, 6.2%, 19.8%, and 0%, respectively while the true positive rates were 81.5%, 91.4%, 98.9%, 93.8%, 80.2%, and 100%, respectively. Compared with normal group, the expression levels of BNIPL and KRT4 were downregulated, while IGFBP3, MMP10, MMP3, and TGFBI were upregulated in LC tissues, which were same as the analysis results in the GEO datasets (Fig. 7A-F).

Receiver operating characteristic curves of hub genes. (A) ROC curves are plotted using expression of BNIPL in GSE84957dataset. (B) ROC curves are plotted using the expression of KRT4 in GSE84957 dataset. (C) ROC curves are plotted using the expression of IGFBP3 in GSE84957 dataset. (D) ROC curves are plotted using the expression of MMP10 in GSE84957 dataset. (E) ROC curves are plotted using the expression of MMP3 in GSE84957 dataset. (F) ROC curves are plotted using the expression of TGFB1 in GSE84957 dataset. The horizontal coordinate is false positive rate and the vertical coordinate is true positive rate, indicating the expression level of the gene in the sample data

The expression of hub genes (BNIPL, KRT4, IGFBP3, MMP10, MMP3, TGFBI) is verified using Gene Expression Profile Interaction Analysis 2 database. (A) BNIPL (B) KRT4 (C) IGFBP3 (D) MMP10 (E) MMP3 (F) TGFBI. *P < 0.05 vs. normal tissues
Analysis of BNIPL in LC
Studies have widely reported that KRT4, IGFBP3, MMP10, MMP3, and TGFBI are involved in the pathogenesis of LC and other cancers [15,16,17,18,19]. There are few reports focusing on the role of BNIPL in cancers. In present study, we selected BNIPL as the target gene for further analysis. Kaplan-Meier survival curve reflected significant differences of survival in patients with high and low expression (Figure. S2). BNIPL expression was significantly correlated with the infiltration of the immune cells in LC (Figure. S3). Low expression level of BNIPL had a significant association with CD8+, CD4+, natural killer cell, and Macrophage, which promoted the immune infiltration (Fig. 8A-D).

Correlation of BNIPL with immune infiltration. (A) Correlation of BNIPL expression with CD8+. (B) Correlation of BNIPL expression with CD4+. (C) Correlation of BNIPL expression with Macrophage. (D) Correlation of BNIPL expression with natural killer cell
Validation of the hub genes expression
qRT-PCR results suggested that the mRNA expression levels of BNIPL and KRT4 in TU212 and TU686 cells were significantly lower than those in NLECs, while the levels of IGFBP3, MMP10, MMP3 and TGFBI were significantly higher than those in NLECs, which was consist with the bioinformatics analysis results (Fig. 9).

The mRNA expression of BNIPL, KRT4, IGFBP3, MMP10, MMP3, and TGFBI in cells. The mRNA expression levels of BNIPL, KRT4, IGFBP3, MMP10, MMP3, and TGFBI in TU212 and TU686 cells were detected by quantitative real time polymerase chain reaction. *P < 0.05, **P < 0.01, ***P < 0.001 vs. normal laryngeal epithelial cells (n = 3)
BNIPL inhibits viability, migration, and invasion of LC cells
The results of transfection efficiency suggested the expression levels of BNIPL in TU212 and TU686 cell lines of pcDNA3.1-BNIPL group were significantly increased, compared to the pcDNA3.1-NC group (P < 0.001) (Fig. 10A). The CCK-8 results suggested that compared with the pcDNA3.1-NC group, the cell viability of TU212 and TU686 cells in pcDNA3.1-BNIPL group was significantly decreased (Fig. 10B). Additionally, the transwell and wound-healing assays revealed that the migration and invasion of TU212 and TU686 cells in the pcDNA3.1-BNIPL group were significantly suppressed, compared with the pcDNA3.1-NC group (Fig. 10C and D).

BNIPL inhibits the viability, migration, and invasion of TU212 and TU686 cells. (A) The transfection efficiency of BNIPL was detected by western blot. (B) The cell viability of TU212 and TU686 cells was evaluated by Cell Counting Kit-8. (C) The migration of TU212 and TU686 cells was assessed by wound-healing assay. (D) The invasion of TU212 and TU686 cells was measured by transwell assay. (Amplification: 200×, Scale: 100 μm). ***P < 0.001 vs. pcDNA3.1-NC group, (n = 3)
