
Abstract
Background
X-linked methyl-CpG-binding protein 2 (MECP2) duplication syndrome is prevalent in approximately 1% of X-linked intellectual disabilities. Accumulating evidence has suggested that MECP2 is the causative gene of MECP2 duplication syndrome. We report a case of a 17-year-old boy with a 1.2 Mb duplication distal to MECP2 on chromosome Xq28. Although this region does not contain MECP2, the clinical features and course of the boy are remarkably similar to those observed in MECP2 duplication syndrome. Recently, case reports have described duplication in the region distal to, and not containing, MECP2. These regions have been classified as the K/L-mediated Xq28 duplication region and int22h1/int22h2-mediated Xq28 duplication region.
The case reports also described signs similar to those of MECP2 duplication syndrome. To the best of our knowledge, ours is the first case to include these two regions.
Case presentation
The boy presented with a mild to moderate regressive intellectual disability and progressive neurological disorder. He developed epilepsy at the age of 6 years and underwent a bilateral equinus foot surgery at 14 years of age because of the increasing spasticity in lower extremities since the age of 11. Intracranial findings showed hypoplasia of the corpus callosum, cerebellum, and brain stem; linear hyperintensity in the deep white matter; and decreased white matter capacity. During his childhood, he suffered from recurrent infection. However, genital problems, skin abnormalities and gastrointestinal manifestations (gastroesophageal reflux) were not observed.
Conclusions
Cases in which duplication was observed in the region of Xq28 that does not include MECP2 also showed symptoms similar to those of MECP2 duplication syndrome. We compared four pathologies: MECP2 duplication syndrome with minimal regions, duplication within the two distal regions without MECP2, and our case including both regions. Our results suggest that MECP2 alone may not explain all symptoms of duplication in the distal part of Xq28.
Similar content being viewed by others


Background
MECP2 is an X-linked transcriptional regulator that binds methylated sites in DNA and regulates the expression of a wide range of genes associated with the nervous system [1]. X-linked MECP2 duplication syndrome, which is prevalent in approximately 1% of X-linked intellectual disabilities, is characterized by infantile hypotonia, severe intellectual disabilities, recurrent infections, progressive spastic disorders, and epilepsy after infancy [2,3,4]. Collins et al. described the possibility that duplications or gain-of-function mutations in MECP2 might underlie some cases of X-linked delayed-onset neurobehavioral disorders [5]. In MECP2 duplication syndrome, different duplication sizes ranging from 0.2–4.0 Mb have been identified [6]. Signs may vary depending on the genes within the region [7], and the role of MECP2 in most of these phenotypes remains unclear.
Duplicated cases in the K/L-mediated Xq28 region (R2: region 2) [8,9,10] and the int22h1/int22h2-mediated Xq28 region (R3: region 3) [11,12,13], which are located distal to MECP2 and do not contain MECP2, have recently been reported. In these case reports, hypotonia, susceptibility to infection, moderate-to-mild intellectual disability, psychiatric signs, and neurological signs were described.
In this report, we describe the case of a 17-year-old Japanese boy with a regressive intellectual disability and progressive neurological disorder. A microarray examination of his DNA identified a 1.2 Mb duplication of maternal origin located 250 kb distal to MECP2 on chromosome Xq28 and containing the two abovementioned regions. The phenotype of this patient was similar to that of MECP2 duplication syndrome, although the duplication occurred in a region not containing MECP2. We compared clinical manifestations in four regions in male-only cases: cases reported as MECP2 duplication syndrome with a particularly small region that does not overlap with other regions (R1: region1) [1, 3, 14], the two regions described above (R2 and R3), and our case. We further discussed why they might present similarly despite not containing MECP2.
Case presentation
The boy (Fig. 1) was born at 38 weeks of gestation with an uncomplicated perinatal course and a birth weight of 2868 g. However, his development has been delayed since infancy. The mother of the patient has borderline intellectual functioning, the maternal uncle showed psychomotor disability, and the father and younger brother were healthy. The patient started raising his head at 3 months of age, crawling at 9 months, walking at 21 months, and speaking at 2 years. The patient was susceptible to infections (respiratory infections or otitis media) and often had fevers. On visiting our hospital at the age of 2 years and 4 months, he presented with a small mouth, fair skin, sacral sinus, flattened foot valgus, muscular hypotonia and resulting constipation, and cervical lymph node swelling. However, hypoplastic genitalia, cryptorchidism, and skin abnormalities were not observed. He spoke only three words, and was diagnosed with a psychomotor disability, and received physical, occupational, and speech therapy. Thyroid function and amino acid and organic acid levels were normal, except for mildly decreased serum IgA levels. He could run at the age of 3 years and speak simple sentences at 3.5 years. At 5 years of age, the Wechsler Intelligence Scale for Children® (WISC)-III indicated a mild intellectual disability, with a total IQ of 63. T1-weighted and fluid-attenuated inversion recovery (FLAIR) images of the magnetic resonance imaging (MRI) of his brain exhibited microcephaly; Verga’s cavity; hypoplasia of the corpus callosum, cerebellum, and brain stem; linear hyperintensity in the deep white matter; and unclear hyperintensity in the cortex and white matter (Fig. 2I).

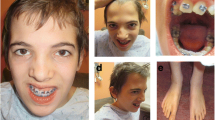
Patient at 7 years and 10 months of age

I Magnetic resonance imaging (MRI) image of patient at 3 years of age: T1-weighted and FLAIR images exhibit microcephaly; Verga’s cavity; hypoplasia of the corpus callosum, cerebellum, and brain stem; linear hyperintensity in the deep white matter; and unclear hypersensitivity in the cortex and white matter. II MRI image at 17 years of age: T1-weighted and FLAIR images show microcephaly; Verga’s cavity; hypoplasia of the corpus callosum, cerebellum, and brain stem; and decreased white matter capacity. The high intensity in the deep white matter observed during infancy was lowered
He developed epilepsy at the age of 6 years and presented with hyperactivity and aggressive behaviors (tantrums and hitting others). Treatment included antiepileptic and psychotropic drugs, such as zonisamide and risperidone, respectively, which helped control epileptic seizures. An electroencephalogram indicated a Θ burst and spike-and-wave connection (lateral view [Rt] > posterior view [Lt]) in the frontal area of the brain (Fig. 3).

Electroencephalogram findings at the age of 6 years revealed a Θ burst and spike-and-wave connection. (Rt > Lt) in the frontal area of the brain
At the age of 10 years, the patient persistently exhibited hyperactivity and aggression, in addition to runaway urge, pain insensitivity, and incontinence; furthermore, he could no longer perform the calculations that he could do previously. The patient also presented with moderate intellectual disability.
At 11 years of age, the spasticity of his lower limbs and equinus varus progressed, and he developed difficulty climbing stairs. Consequently, the patient had undergone bilateral equinus foot surgery at 14 years of age, which enabled him to climb stairs. The fifth edition of the Tanaka-Binet Intelligence Scale suggested moderate intellectual disability, with a total IQ of 39 at 16 years of age. In addition to the previous findings, at 17 years of age, brain MRI findings and T1-weighted and FLAIR images revealed decreased white matter capacity, and the high intensities in this area observed during infancy were lowered (Fig. 2II). Currently, he is enrolled in a special-needs school.
Cytogenetic examinations
Conventional chromosomal G-banding on lymphocytes revealed normal 46, XY karyotype. The FMR1 DNA test results were normal. Subtelomeric rearrangements were excluded using telomere fluorescence in situ hybridization assay (using ToTelVysion Probes, Abbott Molecular Inc., IL 60,007 US). Chromosomal microarray testing using the Agilent 60 K Human Genome Comparative Genomic Hybridization Microarray platform (Agilent Technologies,Santa Clara, CA, USA) revealed approximately 1.2 Mb duplication at Xq28, arr[GRCh37]Xq28(153,558,471 × 1, 153586443_154762275 × 2, 154,783,535 × 2) (Fig. 4), which was inherited from his mother.

Chromosomal microarray testing: arr[hg19] chrX(153,586,397–154,762,275)X2. (q28, gain, 1.2 Mb)
Discussion and conclusions
In the distal Xq28 region, duplication frequently occurs because of the high guanine-cytosine content and low copy repeats [15]. Based on a series of studies reporting patients with genomic duplications within this region, multiple genes/regions are likely responsible for the clinical phenotypes observed in patients harboring different segments of duplication. We compared the clinical manifestations of the four regions: R1, R2, R3, and our case involving both R2 and R3 (Fig. 5; Table 1). The 0.3 Mb R2, 0.5 Mb R3, and our case were distal to MECP2 and did not contain MECP2. The causative gene of our case was within these regions and was presumed to be other than MECP2.

Schematic representation of Xq28, showing the regions of MECP2 duplication syndrome with the minimal regions (R1) and duplication syndromes located distal to MECP2 (R2, R3), and our case
K/L-mediated Xq28 duplication involves 18 genes of which RPL10, ATP6AP1, and GDI1 are highly expressed in the brain, with GDI1 being the most likely causative gene [8, 15]. GDI1 has been identified as X-linked intellectual developmental disorder-41 (XLID41). The increased copy number ofGDI1 correlates with severe neurological signs [7, 9]. The region between int22h1 and int22h2 of Xq28 contains FUNDC2, MTCP1, BRCC3, VBP1, RAB39B, CLIC2, and some genes of F8. The cognitive and neurobehavioral manifestations seen in individuals with MECP2 duplication syndrome are speculated to result from increased dosages of CLIC2 and RAB39B [13]. RAB39B has been identified as X-linked intellectual developmental disorder-72 (XLID72). RAB39B is strongly expressed in the brain and encodes a small GTPase that regulates neuronal development [16]. Vanmarsenille et al. reported that increased doses of RAB39B affects neuronal development and results in a distinct XLID syndrome characterized by autism and cognitive impairment in men [12].
Our patient exhibited spastic diplegia, intellectual disability, post-infant epilepsy, increased susceptibility to infection, and behavioral problems. The spastic neurological symptoms and intellectual regression were progressive. Comparing the symptoms observed in our case with those in men with the other three duplication syndromes led to the following observations:
First, spastic paralysis of both lower limbs starts around puberty, causing difficulty in climbing stairs and requiring surgical intervention. In R2 duplications, 6 of 15 cases (Vandewalle et al. reported three of seven cases [8], Ward et al. reported one of three cases [9], and Sinibald et al. reported two of five cases [10]) exhibited pyramidal tract signs or spasticity (including a description of toe walking that required heel cord lengthening). R2 contains GDI1. It has also been reported that the higher the copy number, the stronger these neurological symptoms, suggesting that GDI1 is the dosage-sensitive gene responsible for these phenotypes [8, 9]. In R3 duplications, only 1 out of 13 case reports exhibited spasticity [12]. In R1 duplications involving MECP2, 7 of 14 cases were reported. Therefore, spasticity occurs more frequently in the duplication of R1 and R2. These areas did not overlap, which may be attributed to the different genetic etiologies in each region. However, progressive spasticity often appears after school age and may not be evident during childhood.
Second, epilepsy can also occur: 3 of the 15 cases with R2 duplications exhibited epileptic phenotypes, whereas none of the 13 cases with R3 duplications had them. In R1 duplications, 10 of the 14 cases reportedly demonstrated epileptic phenotypes. Ward et al. reported that GDI1 mRNA expression in R2 positively correlates with the severity of intellectual deficits [9]. The likelihood of develo** epilepsy is positively correlated with the severity of intellectual deficiency. Cases with R1 duplication presented with severe and intractable epilepsy signs [3, 17], whereas cases with R2 duplication often exhibit relatively mild signs and respond well to medication, as seen in our case.
Third, abnormalities in intracranial findings of the head, including microcephaly, are as follows: In R2 duplications, 11 of 15 cases (Vandewalle et al. reported four of seven cases [8], Ward et al. reported three of three cases [9], and Sinibald et al. reported four of five cases [10]) presented with corpus callosum hypoplasia, and brainstem and cerebellar vermis hypoplasia with ventricular dilatation, suggesting that R2 duplications may cause dysgenesis of the brain during the embryonic period [18]. Yamamoto et al. [7] and Honda et al. [19] stated that duplication of the GDI1 region is associated with hypoplasia of the corpus callosum in patients. In contrast, only 2 (microcephaly and macrocephaly) of 13 cases of R3 duplications has been reported [11,12,13]. In cases with R1 duplications, moderate cerebral atrophy with a hypersignal of the white matter in the posterior parts of the cerebral hemisphere was associated with moderate superior vermis atrophy, moderate cortical atrophy, and a hypersignal of the posterior part of the cerebral white matter. In our case, we observed GDI1-associated findings, such as corpus callosum, brainstem, and cerebellar vermis hypoplasia and abnormally increased hyperintensity of the posterior periventricular cerebral white matter, that are seen in MECP2 syndrome [20, 21].
Fourth, susceptibility to recurrent infections is a unique non-neuropsychiatric feature observed in duplications on chromosome X [22, 23]. In R2 duplications, only 2 of 15 cases exhibited this phenotype. In contrast, recurrent infections were reported in 10 of 13 cases with R3 duplications and 5 of 16 cases with R1 duplications. Therefore, genes within the R1 and R3 regions may be involved in, infection susceptibility. Notably, this phenotype can be overlooked or disregarded during neuropsychiatric evaluation; thus, certain cases may not have been examined in some studies. However, some studies have described associations with genes such as MECP2, IRAK1, and IKBKG within this region [3, 24], although this remains unclear.
Fifth, delayed psychomotor development and intellectual disability are the main features of Xq28 duplication syndrome and were observed in all cases with increased copy numbers in all three regions. MECP2 duplication in R1 was associated with more severe intellectual disability; in R2, there were more moderate cases; and in R3, there were more cases with mild to moderate intellectual disabilities. This suggests that the copy number gain of all three regions causes intellectual disability, with MECP2 effectively having the strongest syndrome, exhibiting even more severe intellectual disability [23]. In contrast, behavioral and neurodevelopmental disorders, including autistic disorders, are not commonly present in patients with duplications in all regions. Neurobehavioral signs, such as attention deficit hyperactivity disorder, impulsivity, and autism spectrum disorder, were present in 9 of 13 cases in the R3 duplicate, whereas only 1 of 15 cases in the R2 duplicate reported behavioral disorders. In R1 duplications, aggressive behavior was reported in only 1 of 13 cases (In R2, F3II.2 by Vandewalle et al. [9] is described as hyperactivity, but this case is excluded because of a perinatal disorder). Thus, neurodevelopmental disorders are particularly common in the int22h1/int22h2-mediated Xq28 duplication syndromes. Increasing RAB39B doses in this region inhibits neuronal development and has been reported to elicit cognitive dysfunction and behavioral problems in patients with a repetitive copy number increase [12, 25]. Mignogna et al. described, using a Rab39b- knockdown mouse model, that the downregulation of RAB39B led to increased GluA2 lacking Ca2 + -permeable aminomethylphosphonic acid receptor composition at the hippocampal neuronal surface and increased dendritic spine density that remained in an immature filopodia-like state [26]. These phenotypes affect behavioral performance in a disease-specific manner. Therefore, we speculate that the most likely causative gene for neurodevelopmental disorders in this region is RAB39B.
In summary, signs such as spasticity of the lower extremities, epilepsy, and brain development failure may involve genes close to MECP2, and behavioral disorders may involve genes in the distal region. Regarding intellectual disabilities, dysfunction in the region containing MECP2 leads to severe cases, whereas dysfunction in the distal region without MECP2 presents mild-to-moderate intellectual disability (Table 2). Since the cases of MECP2 duplication syndrome reported so far vary in size, and many include GDI1 and/or RAB39B, the gene responsible for each symptom remains unclear [27]. In the present study, we confirmed each symptom by comparing it to MECP2 duplication cases that did not include GDI1 or RAB39B. Although each symptom is relatively mild, our case is highly similar to that of the MECP2 syndrome.
We speculated that the causative gene for each sign may not be MECP2 alone. The similarity between the symptoms observed in our case and those in MECP2 duplication syndrome may be because MECP2 regulates the functions of other genes in Xq [4]. Therefore, MECP2 dysfunction can impair the expression of other genes, and the same signs are caused by the dysfunction of each gene. Therefore, the same symptoms may occur due to the dysfunction of different genes. Further studies are necessary to confirm this hypothesis.
Availability of data and materials
The datasets used in the current study are available from the corresponding author upon reasonable request.
Abbreviations
- MECP2 :
-
X-linked methyl-CpG-binding protein 2
- IgA:
-
Immunoglobulin A
- FLAIR :
-
Fluid-attenuated inversion recovery
- MRI:
-
Magnetic resonance imaging
- IQ:
-
Intelligence quotient
- Rt:
-
Right
- Lt:
-
Left
- FMR1 :
-
Fragile X mental retardation1
- RPL10 :
-
Ribosomal protein L10
- ATP6AP1 :
-
ATPase H + transporting accessory protein 1
- GDI1 :
-
GDP dissociation inhibitor 1
- FUNDC2 :
-
FUN14 domain containing 2
- MTCP1 :
-
Mature T cell proliferation 1
- BRCC3 :
-
BRCA1/BRCA2-containing complex subunit 3
- VBP1 :
-
VHL binding protein 1
- RAB39B :
-
Member RAS oncogene family
- CLIC2 :
-
Chloride intracellular channel 2
- F8 :
-
Coagulation factor VIII
- IRAK1 :
-
Interleukin 1 receptor associated kinase 1
- IKBKG :
-
Inhibitor of nuclear factor kappa B kinase regulatory subunit gamma
References
Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9.
Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–53.
Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42: e12.
Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, et al. Structural variation in Xq28: MECP2 duplications in 1 % of patients with unexplained XLMR and in 2 % of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17:444–53.
Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–89.
Peters SU, Fu C, Suter B, Marsh E, Benke TA, Skinner SA, et al. Characterizing the phenotypic effect of Xq28 duplication size in MECP2 duplication syndrome. Clin Genet. 2019;95:575–81.
Yamamoto T, Shimojima K, Shimada S, Yokochi K, Yoshitomi S, Yanagihara K, et al. Clinical impacts of genomic copy number gains at Xq28. Hum Genome Var. 2014;1:14001.
Vandewalle J, Van Esch H, Govaerts K, Verbeeck J, Zweier C, Madrigal I, et al. Dosage-dependent severity of the phenotype in patients with mental retardation due to a recurrent copy-number gain at Xq28 mediated by an unusual recombination. Am J Hum Genet. 2009;85:809–22.
Ward DI, Buckley BA, Leon E, Diaz J, Galegos MF, Hofherr S, et al. Intellectual disability and epilepsy due to the K/L-mediated Xq28 duplication: further evidence of a distinct, dosage-dependent phenotype. Am J Med Genet A. 2018;176:551–9.
Sinibaldi L, Parisi V, Lanciotti S, Fontana P, Kuechler A, Baujat G, et al. Delineation of MidXq28-duplication syndrome distal to MECP2 and proximal to RAB39B genes. Clin Genet. 2019;96:246–53.
El-Hattab AW, Fang P, ** W, Hughes JR, Gibson JB, et al. Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions. J Med Genet. 2011;48:12.
Vanmarsenille L, Giannandrea M, Fieremans N, Verbeeck J, Belet S, Raynaud M, et al. Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum Mutat. 2014;35:377–83.
El-Hattab AW, Schaaf CP, Fang P, Roeder E, Kimonis VE, Church JA, et al. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review. BMC Med Genet. 2015;16:12.
Shimada S, Okamoto N, Ito M, Arai Y, Momosaki K, Togawa M, et al. MECP2 duplication syndrome in both genders. Brain Dev. 2013;35:411–9.
Bauters M, Van Esch H, Friez MJ, Boespflug-Tanguy O, Zenker M, Vianna-Morgante AM, et al. Nonrecurrent MECP2 duplications mediated by genomic architecture driven DNA breaks and break-induced replication repair. Genome Res. 2008;18:847–58.
Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, D’Elia E, et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet. 2010;86:185–95.
Marafi D, Suter B, Schultz R, Glaze D, Pavlik VN, Goldman AM. Spectrum and time course of epilepsy and the associated cognitive decline in MECP2 duplication syndrome. Neurology. 2019;92:108–14.
D’Adamo P, Menegon A, Lo Nigro C, Grasso M, Gulisano M, Tamanini F, et al. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat Genet. 1998;19:134–9.
Honda S, Hayashi S, Nakane T, Imoto I, Kurosawa K, Mizuno S, et al. The incidence of hypoplasia of the corpus callosum in patients with dup (X) (q28) involving MECP2 is associated with the location of distal breakpoints. Am J Med Genet A. 2012;158A:1292–303.
Philippe O, Rio M, Malan V, Van Esch H, Baujat G, Bahi-Buisson N, et al. NF-κB signalling requirement for brain myelin formation is shown by genotype/MRI phenotype correlations in patients with Xq28 duplications. Eur J Hum Genet. 2013;21:195–9.
El Chehadeh S, Faivre L, Mosca-Boidron AL, Malan V, Amiel J, Nizon M, et al. Large national series of patients with Xq28 duplication involving MECP2: delineation of brain MRI abnormalities in 30 affected patients. Am J Med Genet A. 2016;170A:116–29.
Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:1687–95.
Miguet M, Faivre L, Amiel J, Nizon M, Touraine R, Prieur F, et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J Med Genet. 2018;55:359–71.
del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–92.
Zhang W, Ma L, Yang M, Shao Q, Xu J, Lu Z, et al. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020;34:580–97.
Mignogna ML, Ficarella R, Gelmini S, Marzulli L, Ponzi E, Gabellone A, et al. Clinical characterization of a novel RAB39B nonstop mutation in a family with ASD and severe ID causing RAB39B downregulation and study of a Rab39b knock down mouse model. Hum Mol Genet. 2022;31:1389–406.
Andersen EF, Baldwin EE, Ellingwood S, Smith R, Lamb AN. Xq28 duplication overlap** the int22h-1/int22h-2 region and including RAB39B and CLIC2 in a family with intellectual and developmental disability. Am J Med Genet A. 2014;164A:1795–801.
Acknowledgements
We would like to thank the patient’s family for their written informed consent to publish this case report and photographs. We also thank Dr. Noriko Sato and Dr. Masayuki Itoh for their helpful suggestions.
Funding
This research was supported by Intramural Research Grants (2-7, 3-7) for Neurological and Psychiatric Disorders of the National Center of Neurology and Psychiatry (NCNP) in Japan.
Author information
Authors and Affiliations
Contributions
EN, YG, and KI analyzed the patient genetic data on neurological disorders. KI created the schema and contributed to this paper. KA analyzed the patient’s clinical presentation and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Independent Review Board of the National Center of Neurology and Psychiatry, Japan. Written informed consent was obtained.
Consent for publication
Written informed consent to participate was obtained from both parents of the patient. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent for publication of identifying images or other personal or clinical details was obtained from both parents of the patient. We have obtained written consent for (1) genetic analysis using patient specimens and (2) publication of patient photographs.
Competing interests
The authors report no competing interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Akahoshi, K., Nakagawa, E., Goto, Yi. et al. Duplication within two regions distal to MECP2: clinical similarity with MECP2 duplication syndrome. BMC Med Genomics 16, 43 (2023). https://doi.org/10.1186/s12920-023-01465-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01465-3