
Abstract
Background
Geriatric people are prone to suffer from multiple chronic diseases, which can directly or indirectly affect renal function. Through bioinformatics analysis, this study aimed to identify key genes and pathways associated with renal insufficiency in patients with geriatric multimorbidity and explore potential drugs against renal insufficiency.
Methods
The text mining tool Pubmed2Ensembl was used to detect genes associated with the keywords including "Geriatric", "Multimorbidity" and "Renal insufficiency". The GeneCodis program was used to specify Gene Ontology (GO) biological process terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Protein–protein interaction (PPI) networks were constructed using STRING and visualized in Cytoscape. Module analysis was performed using CytoHubba and Molecular Complex Detection (MCODE) plugins. GO and KEGG analysis of gene modules was performed using the Database for Annotation, Visualization and Integrated Discover (DAVID) platform database. Genes clustered in salient modules were selected as core genes. Then, the functions and pathways of core genes were visualized using ClueGO and CluePedia. Finally, the drug-gene interaction database was used to explore drug-gene interactions of the core genes to identify drug candidates for renal insufficiency in patients with geriatric multimorbidity.
Results
Through text mining, 351 genes associated with "Geriatric", "Multimorbidity" and "Renal insufficiency" were identified. A PPI network consisting of 216 nodes and 1087 edges was constructed and CytoHubba was used to sequence the genes. Five gene modules were obtained by MCODE analysis. The 26 genes clustered in module1 were selected as core candidate genes primarily associated with renal insufficiency in patients with geriatric multimorbidity. The HIF-1, PI3K-Akt, MAPK, Rap1, and FoxO signaling pathways were enriched. We found that 21 of the 26 selected genes could be targeted by 34 existing drugs.
Conclusion
This study indicated that CST3, SERPINA1, FN1, PF4, IGF1, KNG1, IL6, VEGFA, ALB, TIMP1, TGFB1, HGF, SERPINE1, APOA1, APOB, FGF23, EGF, APOE, VWF, TF, CP, GAS6, APP, IGFBP3, P4HB, and SPP1 were key genes potentially involved with renal insufficiency in patients with geriatric multimorbidity. In addition, 34 drugs were identified as potential agents for the treatment and management of renal insufficiency.
Similar content being viewed by others
Introduction
With the rapid progress of global aging, the proportion of the geriatric population is gradually increasing, and the growth rate of the population above 80 years old is much higher than that of the population over 65 years old [1]. Geriatric people are prone to suffer from multiple chronic diseases, also known as multimorbidity. The latter refers to the state of having two or more chronic diseases simultaneously [2].
Related studies have revealed that the incidence of multimorbidity in the geriatric population ranges from 55 to 98% [3]. The incidence of renal disease in the geriatric population is much higher than that in the middle and young populations, and the prognosis for the geriatric population is relatively poor [4]. Chronic kidney disease (CKD) generally occurs in the spectrum of geriatric multimorbidity and there are many chronic diseases co-existing with CKD, which are mostly classified as consistent/inconsistent multimorbidity with CKD [5]. Using a single creatinine-based estimating glomerular filtration rate (eGFR) calculation for physiological changes in the body composition that occur with aging does not apply to all adult groups [6]. In the geriatric population, age-related physiological renal changes may lead to a decrease in eGFR. In addition, polypharmacy is common among patients with geriatric multimorbidity due to the presence of multiple diseases. In western countries, the rate of polypharmacy in geriatric multimorbidity patients over 65 years old can reach 30–40% [7]. Therefore, it is imperative to accurately evaluate renal function and identify the pathogenesis of renal insufficiency in geriatric multimorbidity patients to simplify treatment regimens, adjust the dosage and rationalize medication use. Old age is also a factor that interacts with metabolic diseases such as hypertension and diabetes. Therefore, it is critical to evaluate whether renal insufficiency in patients with geriatric multimorbidity is caused by diseases or old age.
Current eGFR assessments in the elderly are mainly derived from equations used to assess eGFR in the young. The 2012 Kidney Disease: Improving Global Outcomes (KDIGO) Clinical Practice Guidelines recommends using the creatinine-derived Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation to estimate eGFR in routine practice. The guidelines suggest that cystatin C-based equations have the potential to improve the diagnosis and epidemiology of CKD and should only be used in individuals with an eGFR between 45 and 59 ml/min/1.73 m2 with no other evidence of CKD [8], suggests limited availability of eGFR assessments rather than poor performance. Assessing the eGFR by measuring biomarkers in urine or blood is invasive and time-consuming. There has been significant progress in using transdermal measurements of eGFR in murine models [9, 10], which are non-invasive and allow real-time measurement of eGFR. Transdermal measurement of eGFR may be the key to the non-invasive and real-time measurement of eGFR in various clinical settings in the future.
At present, multiple studies have established that the interaction between metabolic diseases and aging affects renal function in older people and the underlying pathogenic cause is cellular dysfunction. A meta-analysis of genome-wide association studies found that the genes identified in the eGFR locus are highly expressed in renal tissues and pathways associated with renal development, transmembrane transporter activity, renal structure, and the regulation of glucose metabolism [11]. Metabolic analysis in CKD animal models also revealed alterations in numerous metabolites such as metabolic reprogramming of the S-nitroso-coa reductase system that can prevent renal insufficiency. The regulation of PKM2 in the mouse proximal tubules may be a new perspective in the treatment of renal insufficiency [12]. At the cellular level, it has also been observed that blood glucose and albumin affect the cellular metabolic expression of four kinds of innate cells in the kidneys [13]. For patients with moderate to high risk of CKD, bariatric surgery can maintain better levels of eGFR in obese patients, suggesting that the incidence of renal disease can be reduced by improving metabolic risk [14]. It has also been determined that blood homocysteine level in the elders is closely related to age-induced renal insufficiency [15], suggesting that renal insufficiency in older people is related to cell metabolism.
Geriatric multimorbidity complicated with renal insufficiency can influence the prognosis of patients. However, most of the above studies have assessed the risk of renal disease development in a certain disease, and there is still a lack of tools to accurately assess the impact of multimorbidity on renal insufficiency. Clinicians should precisely identify renal insufficiency in geriatric multimorbidity patients to provide an early diagnosis and treatment to slow down the progression and prevent or delay end-stage renal disease (ESRD). Discovering novel pharmacotherapy by traditional means can be time-consuming and expensive, while treating diseases beyond their original development purpose through drug reuse may be more effective and faster [16]. New information about old drugs and new therapies can be obtained through the text mining of bioinformatics [17]. The purpose of this study was to explore the existing published literature and biological databases and use other analysis tools for the assessment of renal insufficiency in geriatric multimorbidity patients, to further clarify the molecular mechanism of renal insufficiency in geriatric patients with multimorbidity and identify potential therapeutic targets to guide rational clinical drug use better.
Methods and materials
Text mining
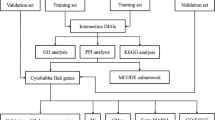
Text mining was conducted using Pubmed2Ensembl (http://www.pubmed2ensembl.org), which is an extension of the BioMart system. It links more than 2 million articles in PubMed to approximately 150,000 genes from 50 species in Ensembl [18]. We entered "Geriatric" and "Multimorbidity" in the search box in Pubmed2Ensembl. Then non-duplicate genes were extracted. The union of the extracted genes from the two gene sets was defined as the gene set associated with "Geriatric Multimorbidity" and was denoted as the "G-M" gene set. We entered the search term "Renal insufficiency " in Pubmed2Ensembl to extract the unrepeated genes. After retrieving the related genes, the "G-M" and "Renal insufficiency" genes sets were intersected using Venny (https://bioinfogp.cnb.csic.es/tools/venny/). The intersection of genes extracted from two gene sets constituted our text mining genes (TMGs).
Biological process and pathway enrichment analysis of TMGs
Genecodis is a powerful web-based tool for the functional interpretation of genomics experimental technical results, which integrates different sources of information to search for annotations that often coexist in a set of genes and ranks them based on statistical significance [19]. The TMGs from our text mining were entered and analyzed using the GO biological process analogy. Then, significantly enriched biological process genes were selected. The enriched genes were further analyzed using the KEGG pathways annotation. Afterward, the genes involved in the significantly enriched KEGG pathways were selected for further analysis.
Integration of protein–protein interaction (PPI) network and identification of Hub genes
Results obtained from the gene enrichment analysis in the previous step were applied to the gene retrieval tool STRING (http://string-db.org) for PPI analysis [20]. STRING provides a platform that can analyze PubMed text mining data and integrate multiple database resources. It covers about 24.6 million proteins from 5090 organisms and more than 3.1 billion interactions, which can be used to analyze the relationships and interactions between proteins. First, STRING was employed to construct the PPI network of different genes. In addition, the application of CytoHubba in Cytoscape was used to identify hub genes [21]. CytoHubba for hub Genes is a Cytoscape plugin that provides 11 topological analysis methods to sort nodes in a network based on the network characteristics [22]. According to the results of correlation analysis, the relevant genes with a degree ≥ 10 were screened out for further analysis.
Molecular Complex Detection (MCODE) analysis of subnet networks
MCODE constructed by Cytoscape was downloaded and ran using a Cytoscape visualization network of molecular interaction to screen important gene modules in the visual network of molecular interaction [23]. In the analysis results, the gene modules in the analysis were sorted according to the network score. The gene module with the highest score was an important gene module in the visual network of molecular interaction screening, which represented the most critical and typical genes in the network. Two gene modules (including 40 genes) with the highest network score from the PPI network were selected for further verification and analysis.
Gene ontology and KEGG pathway enrichment analysis of module genes
The GO functional and KEGG pathway enrichment analysis of important module genes was performed using the online gene function analysis tool DAVID (https://david.ncifcrf.gov/) [24]. DAVID is a database of annotation, visualization and integrated discovery of biological information, which can associate the genes in the input list to the term of biological annotation and find the most significantly enriched biological annotation through statistical methods. At present, it is mainly used for related function and pathway enrichment analysis of differential genes. The FDR we chose in the DAVID analysis uses the Benjamin-Hochberg multiple test method to approximate control the error detection rate [25]. Before Gene ontology and KEGG pathway enrichment analysis, we set FDR < 0.05 was the cut-off point. GO (http://www.geneontology.org) database contains three main categories of genome data function classification terms including biological process, cell composition and molecular work [26]. KEGG (www.genome.jp/kegg) is a knowledge base for systematic analysis, annotation and visualization of gene function [27]. We set the Cytoscape plugins ClueGo and Cluepedia to P < 0.01 was considered statistically significant to visualize the GO function of core genes and KEGG pathway enrichment analysis [28].
Drug-gene interactions
The drug-gene interaction database (DGIDB) mines available resources to generate hypotheses about how mutated genes can be targeted for therapy or prioritized for drug development [29]. It provides information on drug-gene interactions and known or potential drug associations to genes. DGIDB was utilized to explore drug-gene interactions associated with the identified genes to generate potential targets for existing drugs or compounds.
Results
Acquisition of TMGs
Based on the text mining strategy described in the Methods and Materials section (Fig. 1), 1046 unique genes were related to the geriatric population, 18 unique genes were related to multimorbidity, and 684 unique genes were related to renal insufficiency. Then, all non-duplicate genes were extracted. The union of the extracted genes from the two gene sets associated with geriatric multimorbidity was denoted as the G-M gene set. Among them, 351 genes overlapped between the G-M and the renal insufficiency set. Hence, we considered that those 351 genes participated in the biological processes of renal insufficiency in patients with geriatric multimorbidity (Fig. 2A).

Summary of the whole study design. Text mining procedures were conducted using Pubmed2Ensembl to identify genes related to Geriatric Multimorbidity (G-M) and Renal insufficiency. Genes collection enrichment was used GeneCodis to find genes enriched in the GO biological process terms and KEGG pathways. STRING and CytoHubba were used to construct a protein–protein interaction network and screen the proteins encoded by the hub genes according to the degree of the nodes. MCODE were used to identify the related protein network modules and calculate the score of each module. DAVID and ClueGO were used to analyze the GO biological process terms and KEGG pathways. DGIDB was used to classify potential drug targets based on the lists of significant genes. KEGG: Kyoto Encyclopedia of Genes and Genomes

Identification and enrichment analysis of the TMGs. A Venn diagram analysis was carried out between the G-M and Renal insufficiency using the Venny website. The 351 genes that were common were considered to be associated to G-M and Renal insufficiency. B The protein–protein interaction (PPI) network of the 216 target TMGs were visualized by Cytoscape
Gene ontology and KEGG pathway analysis
The GeneCodis website was used to visualize GO functional and KEGG pathways to determine the most enriched terms closely related to renal insufficiency in patients with geriatric multimorbidity. The result of our analysis showed that 2717 significantly enriched GO biological process annotations were identified as 343 unique genes. Among them, the five most enriched terms were "cytokine-mediated signaling pathway" (P = 2.02245E−57), "signal transduction" (P = 1.40787E−45), "negative regulation of apoptotic process" (P = 5.31297E−44), "positive regulation of gene expression" (P = 4.56741E−41) and "response to drug" (P = 5.19875E−39), for 52, 77, 51, 48 and 39 TMGs, respectively (Table 1). Other highly enriched biological processes included "positive regulation of transcription by RNA polymerase II", "inflammatory response", "response to lipopolysaccharide", "cellular protein metabolic process" and "response to hypoxia".
To study the function of TMGs and the enrichment of signaling pathways, KEGG pathway enrichment analysis was also performed on the identified TMGs using GeneCodis. The KEGG pathway enrichment analysis identified 242 important pathways involving 270 TMGs. Table 2 shows that most of the important signaling pathways that were enriched were "pathways in cancer" (P = 2.40582E−63), "cytokine-cytokine receptor interaction" (P = 9.03679E−52), "AGE-RAGE signaling pathways in diabetic complications" (P = 1.20609E−43), "PI3K-Akt signaling pathway" (P = 1.90881E−39) and "HIF-1 signaling pathway " (P = 6.07415E−36) involved 58, 39, 29, 36 and 25 TMGs, respectively (Table 2). Other highly concentrated pathways included the “metabolic pathways”, “proteoglycans in cancer”, “transcriptional misregulation in cancer”, “fluid shear stress and atherosclerosis” and "Human cytomegalovirus infection".
PPI network construction, identification of Hub genes and Modular analysis
Various biological data networks can be obtained through correlation analysis, including information about signal transduction pathways, gene regulation and protein–protein interaction. We input the target genes into the STRING website. A PPI network of 270 target genes was constructed through analysis. The constructed PPI network consisted of 216 nodes and 1087 edges (Fig. 2B). Five of those genes were not included in the constructed PPI network. We ran the plugin CytoHubba in Cytoscape. The topological network algorithm was used to assign a value to each gene and rank each gene according to its degree in the correlation analysis result. The darker the color, the higher the score and the more significant the gene (Fig. 3A). According to the analysis results, 79 nodal genes with nodal degree ≥ 10 were selected (Table 3). Then, we found the hub genes in the PPI network, which referred to the gene with the highest connection in the module. As illustrated in Fig. 3B, APP, IL6, KNG1, AKT1, VEGFA, APOB, FN1, TIMP1, ALB and TNF were the 10 hub genes with the highest connectivity in the module. If these selected genes were supported by relevant clinical research data in the research progress, we could further study the association between these proteins and patient survival. To further obtain more gene modules, we used the MCODE plugin to analyze the 79 target genes screened in the previous step and obtained 5 modules (Fig. 4A–E). We selected the two modules with the highest scores (including 40 genes) by calculating network scores for further analysis. Module 1 contained 26 nodes and 241 edges. Module 2 contained 14 nodes and 69 edges.

Categorize the degree and analyze Hub genes. A All nodes of PPI were presented according to degree by CytoHubba. The degree decreases from inside to outside and the color changes from dark to light. B The first 10 hub genes in the macro module were identified by CytoHubba plug-in. The image shows degree from red to yellow, the significance of genes declines

Description and enrichment analysis of the TMGs. A–E The five modules were carried out from PPI network using MCODE. A Module 1, the most significant module with 26 nodes; B Module 2; C Module 3; D Module 4; E Module 5
In order to further analyze the enrichment of core genes, we conducted Gene ontology and KEGG pathway analysis for the two modules selected in the previous step. The results showed that the 26 genes in module 1 were mainly related to platelet degranulation (BP), cytoplasmic membrane-bounded vesicle lumen (CC) and receptor binding (MF) (Fig. 5A). On the other hand, the 14 genes in module 2 were mainly associated with G-protein coupled receptor signaling pathway (BP), extracellular space (CC), and G-protein coupled receptor binding (MF) (Fig. 5B). Pathway enrichment analysis showed that the genes in module 1 were associated with the HIF-1, PI3K-Akt, MAPK, Rap1, and FoxO signaling pathways (Fig. 5C). Conversely, the genes in module 2 were significantly correlated with the chemokine, phospholipase, and Nod-like receptor signaling pathways (Fig. 5D). Module 1 contained 26 genes with 241 edges, all of which were core genes, indicating that module 1 played a vital role in the PPI network. 26 genes were selected as core candidate genes for the PPI networks. The enrichment analysis indicated that these genes were significantly enriched in platelet alpha granule lumen, HIF-1 signaling pathway, Melanoma, protein kinase activator activity, Complement and coagulation cascades and AGE-RAGE signaling pathway in diabetic complications (P < 0.01, Fig. 6).

Gene ontology and KEGG pathway analysis of the genes in the first two modules. A Top 18 significantly enriched GO terms in module 1. B Top 12 Significantly enriched GO terms in module 2. C Top 15 significantly enriched KEGG pathways in module 1. D Top 15 significantly enriched KEGG pathways in module 2. The functional and pathway enrichment analyses were performed using DAVID. KEGG: Kyoto Encyclopedia of Genes and Genomes

Function analysis of the 26 core genes in module 1. A Enriched GO terms and KEGG pathways. B Functions and pathways of the core genes were computed and visualized using ClueGO. C Distribution of the functions and pathways among the core genes. Each function or pathway is color coded. Corrected P < 0.01 was considered statistically significant. KEGG: Kyoto Encyclopedia of Genes and Genomes
Drug-gene interaction analysis of core genes
The final confirmed genes were used to conduct drug-gene interaction analysis and an initial list of 34 drugs was obtained (Table 4). Thirty-four drugs targeted by 21 of the 26 core genes (except IGF1, KNG1, PF4, SERPINA1 and TIMP1) may be potential therapeutic agents for renal insufficiency in geriatric multimorbidity patients. The 26 core genes in the analysis results were mainly enriched in PI3K-Akt signaling Pathway, HIF-1 signaling Pathway, Pathways in cancer and Vitamin digestion and absorption. The main links between drugs, genes, and pathways are displayed in Fig. 7.

Sankey diagram display the essential connections among drugs, genes, and pathways. Drug-gene interactions were analyzed for 26 genes in module 1. We found 21 genes that target 34 potential existing drugs. In addition, these 21 genes are mainly enriched in 4 KEGG pathways. "None" in the pathway means that the core gene has no related pathway
Discussion
By applying a series of bioinformatics methods to explore gene expression, we identified 351 genes involved in renal insufficiency in geriatric multimorbidity patients. Among these genes enriched go biological process terms, which have so far been shown to be primarily associated with renal insufficiency iucluding cytokine-mediated signaling pathway [30], signal transduction [31], negative regulation of apoptotic process [32], positive regulation of gene expression [33], response to drugs [34], positive regulation Of transcription by RNA polymerase II [35], inflammatory response [36], response to lipopolysaccharide [37], cellular protein metabolic process [38] and response to hypoxia [39]. In addition, the results of KEGG pathway analysis corresponding to 351 genes related to renal insufficiency including cytokine-cytokine receptor interaction [40], AGE-RAGE signaling pathways in diabetic complications [41], PI3K-Akt signaling pathway [42], HIF-1 signaling pathway [42], metabolic pathways [43], proteoglycans in cancer [44], transcriptional misregulation in cancer [45], fluid shear stress and atherosclerosis [46] and Human cytomegalovirus infection [47]. Pathways in cancer may be associated with renal impairment, but the correlation needs to be further verified.
The PPI network and CytoHubba explored the correlation connectivity between these genes (Fig. 3A) and identified 10 pivotal genes, including APP, IL6, KNG1, AKT1, VEGFA, APOB, FN1, TIMP1, ALB, and TNF(Fig. 3B). Relevant literature have shown that IL6, AKT1, VEGFA, FN1, TIMP1, ALB and TNF are associated with renal insufficiency. According to current literature, there are no studies on the other hub genes and their effect on renal insufficiency. However, these genes may also play a significant role in renal insufficiency. The MCODE analysis generated 26 core genes, including CST3, SERPINA1, FN1, PF4, IGF1, KNG1, IL6, VEGFA, ALB, TIMP1, TGFB1, HGF, SERPINE1, APOA1, APOB, FGF23, EGF, APOE, VWF, TF, CP, GAS6, APP, IGFBP3, P4HB, and SPP1, that participated in the HIF-1, PI3K-Akt, MAPK, Rap, and FoxO signaling pathways.
The results showed that Interleukin 6 (IL-6) is an autocrine growth factor secreted by mesangial cells and is involved in the pathological proliferation of mesangial cells [48]. A meta-analysis showed that serum cystatin C is superior to serum creatinine as a marker of renal function [49]. Serum albumin levels have been shown to further predict the clinical outcomes in patients with CKD undergoing cardiac resynchronization therapy [50]. Among these genes, APOA1, APOB, and APOE all belong to the apolipoprotein family. ApoA1 was negatively associated with eGFR decline during a short period of one year [51]. APOB-containing lipoproteins complex of the medium and low-density apolipoprotein complexes may contribute to renal insufficiency by interacting with glomerular or tubulointerstitial problems [52]. Patients with high APOE2 levels have a higher risk of develo** CKD or even ESRD because APOE2 affects the clearance of very-low-density lipoprotein (VLDL) and chylomicron (CM) remnants [53]. FN1 mutations lead to glomerular disease with fibronectin deposition [54]. Excessive FN production can accelerate glomerulosclerosis and tubulointerstitial fibrosis and increase the incidence of diabetic nephropathy (DN) by causing the thickening of the glomerular and tubular basement membranes [55]. Fibroblast growth factor 23 (FGF23) regulates phosphate reabsorption and 1alpha-hydroxylase activity in the kidney [56]. Studies have shown that FGF23 levels appear to be independently associated with mortality in dialysis patients [57]. SPP1 (Osteopontin) is a cell-attached glycoprotein and its expression correlated with the severity of the renal tubulointerstitial injury [58]. In the process of renal aging, the high expression of TIMP-1 up-regulates the expression of PTEN through an MMP-independent pathway and subsequently leads to aging-related vascular damage [59]. TGFB1 can further induce renal interstitial fibrosis through endothelial-to-mesenchymal transition (EndMT) [60]. Individuals with renal insufficiency with increased VWF levels may have an increased risk of venous thrombosis [61]. Decreased vascular endothelial growth factor A (VEGFA) levels are associated with glomerular microangiopathy [62]. Currently, there are few studies on the direct relationship between APP, CP, EGF, GAS6, HGF, IGF1, IGFBP3, KNG1, SERPINE1, SERPINA1, TF, PF4 and P4HB, and renal insufficiency in relevant literature, which can provide a reference for future studies on renal insufficiency with geriatric multimorbidity patients.
HIF is involved in the renal fibrosis process during the disease course of CKD through gene transcription, signaling pathways, epithelial-mesenchymal transition and epigenetic regulation [63]. The PI3K-Akt signaling pathway is mainly involved in regulating cell proliferation, migration, differentiation and angiogenesis [64]. Helix B surface peptide (HBSP) can improve renal ischemia–reperfusion injury, renal function and also improve apoptosis after ischemia–reperfusion injury by regulating the PI3k/Akt pathway [65]. Activating mitogen-activated protein kinase (MAPK) and lipopolysaccharide (LPS) can lead to the increased transcription of pro-inflammatory cytokines, which can directly affect renal parenchyma, promote renal tubular cell apoptosis and directly induce AKI [66]. Decreased RAP1-GTP mediated by elevated RAP1GAP levels may be a crucial factor in inducing podocyte dysfunction in human glomerular disease [67]. FoxO bound to β-catenin can prevent rHTGF-1-induced profibrosis [68].
As many drugs are metabolized through the kidneys, they might damage renal function. Potential drugs identified through the search of drug-gene interaction are mainly divided into protecting renal function, damaging renal function and adjusting doses according to renal function. Burosumab treats x-linked patients with hypophosphatemia (XLH) by antagonizing FGF23, increasing renal tubular phosphate reabsorption and normalizing serum phosphorus concentrations [69]. Additional preclinical and clinical studies are warranted to determine whether blocking FGF23 with burosumab will provide a new targeted intervention for mineral metabolism disorders in CKD [70]. Adalimumab (ADA) is a tumor necrosis factor (TNF-α) inhibitor that reduces or suppresses inflammatory processes by inhibiting pro-inflammatory cytokines [71]. ADA pretreatment may play a role in experimental renal insufficiency [72]. Siltuximab inhibited tumor growth of human renal carcinoma in nude mice by binding to IL-6 [73]. Related studies have shown that angiotensin receptor blockers (ARB) can play a role in renal protection through multiple and complex mechanisms [74]. Olmesartan may delay or prevent microalbuminuria in type 2 diabetes mice, improving renal function [75]. In animal models, ramipril significantly reduced the number of glomerular and tubule-interstitial fibrosis and activated fibroblasts, exerting a protective effect on the kidney [76]. Defibrotide can prevent the upregulation of endothelial dysfunction markers induced by a uremic environment, and downregulate the expression of HDACs through the PI3/AKT signaling pathway, thus playing a protective role in the endothelia [77]. In addition, cholestyramine [78], mipomersen [79] and soybean oil can regulate lipid metabolism and directly improve renal function [80]. Glp-1 RA inhibits NHE3-dependent sodium reabsorption in the proximal tubule and has a direct renal protective effect on the renin-angiotensin system, which can improve inflammation, ischemia/hypoxia, apoptosis and neural signaling [81]. Studies have shown that imatinib mesylate interferes with PDGF and TGF-β activated signaling cascade and improves renal tubulointerstitial fibrosis [82]. Hydroxychloroquine (HCQ) reduces renal insufficiency by downregulating NLRP3 inflammasomes activation mediated by CTSB and CTSL [83].
According to literature, ranibizuma [84], cetuximab [85], aflibercept [85], warfarin [86], gadofosveset [87], fluorouracil [88] and penicillamine [89] may cause renal insufficiency through a variety of ways. Moreover, the dose of ribavirin [90], ganciclovir [91], metronidazole [92], levofloxacin [93] and digoxin [94] needs to be adjusted according to renal function. Currently, the relationship between ademetionine, caplacizumab, pegaptanib sodium, lomitapide mesylate, calcitonin, urokinase, cetrorelix, iodipamide, dexrazoxane, ocriplasmin, and renal insufficiency has not been reported in the existing literature. Future studies could further investigate the association between these drugs and renal insufficiency.
In conclusion, we identified 26 core genes, CST3, SERPINA1, FN1, PF4, IGF1, KNG1, IL6, VEGFA, ALB, TIMP1, TGFB1, HGF, SERPINE1, APOA1, APOB, FGF23, EGF, APOE, VWF, TF, CP, GAS6, APP, IGFBP3, P4HB, and SPP1, that may be related to renal insufficiency in patients with geriatric multimorbidity. These genes were enriched in the HIF-1, PI3K-Akt, MAPK, Rap1, and FoxO signaling pathways. We also identified 34 drugs that may help guide the future treatment of renal insufficiency in patients with geriatric multimorbidity. The lack of experimental verification is a limitation of this study, and further experimental studies are needed to verify these results.
Availability of data and materials
The authors declare that the data supporting the findings of this study are available within the article. The datasets analysed during the current study are publicly available in the pubmed2ensembl (http://www.pubmed2ensembl.org).
Abbreviations
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MCODE:
-
Molecular Complex Detection
- DAVID:
-
Database for Annotation, Visualization and Integrated Discover
- CKD:
-
Chronic kidney disease
- eGFR:
-
Estimating glomerular filtration rate
- KDIGO:
-
Kidney Disease: Improving Global Outcomes
- CKD-EPI:
-
Chronic Kidney Disease Epidemiology Collaboration
- ESRD:
-
End-stage renal disease
- PPI:
-
Protein–protein interaction
- DGIDB:
-
Drug-gene interaction database
- VLDL:
-
Very-low-density lipoprotein
- CM:
-
Chylomicron
- DN:
-
Diabetic nephropathy
- FGF23:
-
Fibroblast growth factor 23
- EndMT:
-
Endothelial-to-mesenchymal transition
- VEGFA:
-
Vascular endothelial growth factor A
References
Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374(9696):1196–208.
Tinetti ME, Fried TR, Boyd CM. Designing health care for the most common chronic condition–multimorbidity. JAMA. 2012;307(23):2493–4.
Marengoni A, Angleman S, Melis R, et al. Aging with multimorbidity: a systematic review of the literature. Ageing Res Rev. 2011;10(4):430–9.
Wang F, Yang C, Long J, et al. Executive summary for the 2015 annual data report of the China Kidney Disease Network (CK-NET). Kidney Int. 2015;95(3):501–5.
Bowling CB, Plantinga L, Phillips LS, et al. Association of multimorbidity with mortality and healthcare utilization in chronic kidney disease. J Am Geriatr Soc. 2017;65(4):704–11.
Raman M, Middleton RJ, Kalra PA, Green D. Estimating renal function in old people: an in-depth review. Int Urol Nephrol. 2017;49(11):1979–88.
Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999–2012. JAMA. 2015;314(17):1818–31.
Andrassy KM. Comments on ‘KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease.’ Kidney Int. 2013;84(3):622–3.
Schock-Kusch D, Sadick M, Henninger N, et al. Transcutaneous measurement of glomerular filtration rate using FITC-sinistrin in rats. Nephrol Dial Transpl. 2009;24(10):2997–3001.
Schreiber A, Shulhevich Y, Geraci S, et al. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol. 2012;303(5):F783–8.
Pattaro C, Teumer A, Gorski M, et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat Commun. 2016;7:10023.
Zhou HL, Zhang R, Anand P, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature. 2019;565(7737):96–100.
Lagies S, Pichler R, Bork T, et al. Impact of diabetic stress conditions on renal cell metabolome. Cells. 2019;8(10):1141.
Lin YC, Lai YJ, Lin YC, et al. Effect of weight loss on the estimated glomerular filtration rates of obese patients at risk of chronic kidney disease: the RIGOR-TMU study. J Cachexia Sarcopenia Muscle. 2019;10(4):756–66.
Ostrakhovitch EA, Tabibzadeh S. Homocysteine and age-associated disorders. Ageing Res Rev. 2019;49:144–64.
Moosavinasab S, Patterson J, Strouse R, et al. ‘RE:fine drugs’: an interactive dashboard to access drug repurposing opportunities. Database (Oxford). 2016;2016:083.
Gauthier J, Vincent AT, Charette SJ, Derome N. A brief history of bioinformatics. Brief Bioinform. 2019;20(6):1981–96.
Zhou J, Fu BQ. The research on gene-disease association based on text-mining of PubMed. BMC Bioinform. 2018;19(1):37.
Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012;40:W478–83.
Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. 2019;18(2):623–32.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Bandettini WP, Kellman P, Mancini C, et al. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: a clinical validation study. J Cardiovasc Magn Reson. 2012;14(1):83.
Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a new and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Alterovitz G, **ang M, Mohan M, Ramoni MF. GO PaD: the Gene Ontology Partition Database. Nucleic Acids Res. 2007;35:D322–7.
Kanehisa MFM, Tanabe MSY, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–61.
Bindea G, Galon J, Mlecnik B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29(5):661–3.
Freshour SL, Kiwala S, Cotto KC, et al. Integration of the drug-gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021;49(D1):D1144–51.
Roubicek T, Bartlova M, Krajickova J, et al. Increased production of proinflammatory cytokines in adipose tissue of patients with end-stage renal disease. Nutrition. 2009;25(7–8):762–8.
Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm–from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9(10):587–98.
Zhao XC, Livingston MJ, Liang XL, Dong Z. Cell apoptosis and autophagy in renal fibrosis. Adv Exp Med Biol. 2019;1165:557–84.
Shi H, Yu J, Li J. Nephroblastoma overexpressed gene expression and its prognostic implications of clinical outcomes in renal cell carcinoma patients. Cancer Biomark. 2017;20(3):241–6.
Ashley C. Retraction notice. JAAD Case Rep. 2018;4(8):841.
Wang W, Hu W, Wang Y, An Y, Song L, Shang P, et al. Long non-coding RNA UCA1 promotes malignant phenotypes of renal cancer cells by modulating the miR-182-5p/DLL4 axis as a ceRNA. Mol Cancer. 2020;19(1):18.
Cobo G, Lindholm B, Stenvinkel P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol Dial Transpl. 2018;33(suppl_3):iii35–40.
Lu XM, Ma L, ** YN, Yu YQ. Lumican overexpression exacerbates lipopolysaccharide-induced renal injury in mice. Mol Med Rep. 2015;12(3):4089–94.
Rajani R, Pastor-Soler NM, Hallows KR. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr Opin Nephrol Hypertens. 2017;26(5):375–83.
Liu M, Ning X, Li R, et al. Signalling pathways involved in hypoxia-induced renal fibrosis. J Cell Mol Med. 2017;21(7):1248–59.
Zeng Q, Sun S, Li Y, Li X, Li Z, Liang H. Identification of therapeutic targets and prognostic biomarkers among CXC chemokines in the renal cell carcinoma microenvironment. Front Oncol. 2019;9:1555.
Tuleta I, Frangogiannis NG. Diabetic fibrosis. Biochim Biophys Acta Mol Basis Dis. 2021;1867(4): 166044.
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45(8):860–7.
Grayson PC, Eddy S, Taroni JN, Lightfoot YL, Mariani L, Parikh H, et al. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis. 2018;77(8):1226–33.
Niedworok C, Kempkensteffen C, Eisenhardt A, Tschirdewahn S, Rehme C, Panic A, et al. Serum and tissue syndecan-1 levels in renal cell carcinoma. Transl Androl Urol. 2020;9(3):1167–76.
Karpova Y, Guo D, Makhov P, Haines AM, Markov DA, Kolenko V, et al. Poly(ADP)-ribosylation inhibition: a promising approach for clear cell renal cell carcinoma therapy. Cancers (Basel). 2021;13(19):4973.
Malone A, Chari D, Cournane S, Naydenova I, Fagan A, Browne J. Investigation of the assessment of low degree (<50%) renal artery stenosis based on velocity flow profile analysis using Doppler ultrasound: an in-vitro study. Phys Med. 2019;65:209–18.
López Giuliani AC, Hernández E, Tohmé MJ, Taisne C, Roldán JS, García Samartino C, et al. Human cytomegalovirus inhibits autophagy of renal tubular epithelial cells and promotes cellular enlargement. Front Cell Infect Microbiol. 2020;10:474.
Ruef C, Budde K, Lacy J, et al. Interleukin 6 is an autocrine growth factor for mesangial cells. Kidney Int. 1990;38(2):249–57.
Dharnidharka VR, Kwon C, Stevens G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta-analysis. Am J Kidney Dis. 2002;40(2):221–6.
Uchikawa T, Shimano M, Inden Y, Murohara T. Serum albumin levels predict clinical outcomes in chronic kidney disease (CKD) patients undergoing cardiac resynchronization therapy. Intern Med. 2014;53(6):555–61.
Hayashi K, Takayama M, Abe T, et al. Investigation of Metabolic Factors Associated with eGFR Decline Over 1 Year in a Japanese Population without CKD. J Atheroscler Thromb. 2017;24(8):863–75.
Havel RJ. Role of triglyceride-rich lipoproteins in progression of atherosclerosis. Circulation. 1990;81(2):694–6.
Eto M, Saito M, Okada M, et al. Apolipoprotein E genetic polymorphism, remnant lipoproteins, and nephropathy in type 2 diabetic patients. Am J Kidney Dis. 2002;40(2):243–51.
Castelletti F, Donadelli R, Banterla F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A. 2008;105(7):2538–43.
Gong W, Li J, Chen Z, et al. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating CKIP-1 to resist HG-induced up-regulation of FN and ICAM-1 in GMCs and diabetic mice kidneys. Free Radic Biol Med. 2017;106:393–405.
Stubbs JR, Liu S, Tang W, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol. 2007;18(7):2116–24.
Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–92.
Pichler R, Giachelli CM, Lombardi D, et al. Tubulointerstitial disease in glomerulonephritis. Potential role of osteopontin (uropontin). Am J Pathol. 1994;144(5):915–26.
Chen JX, Cai GY, Chen XM, et al. Effect of TIMP1 transfection on PTEN expression in human kidney proximal tubular cells. Genet Mol Res. 2015;14(4):17373–83.
Wang Z, Han Z, Tao J, et al. Role of endothelial-to-mesenchymal transition induced by TGF-β1 in transplant kidney interstitial fibrosis. J Cell Mol Med. 2017;21(10):2359–69.
Ocak G, Vossen CY, Lijfering WM, et al. Role of hemostatic factors on the risk of venous thrombosis in people with impaired kidney function. Circulation. 2014;129(6):683–91.
Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol. 2020;16(5):289–303.
Liu J, Wei Q, Guo C, et al. Hypoxia, HIF, and associated signaling networks in chronic kidney disease. Int J Mol Sci. 2017;18(5):0950.
Zhang H, Bajraszewski N, Wu E, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117(3):730–8.
Yang C, Zhao T, Lin M, et al. Helix B surface peptide administered after insult of ischemia reperfusion improved renal function, structure and apoptosis through beta common receptor/erythropoietin receptor and PI3K/Akt pathway in a murine model. Exp Biol Med (Maywood). 2013;238(1):111–9.
Ren Q, Guo F, Tao S, Huang R, Ma L, Fu P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomed Pharmacother. 2020;122: 109772.
Potla U, Ni J, Vadaparampil J, et al. Podocyte-specific RAP1GAP expression contributes to focal segmental glomerulosclerosis-associated glomerular injury. J Clin Invest. 2014;124(4):1757–69.
Rao P, Pang M, Qiao X, et al. Promotion of β-catenin/Foxo1 signaling ameliorates renal interstitial fibrosis. Lab Invest. 2019;99(11):1689–701.
Carpenter TO, Whyte MP, Imel EA, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378(21):1987–98.
Perwad F, Portale AA. Burosumab therapy for X-linked hypophosphatemia and therapeutic implications for CKD. Clin J Am Soc Nephrol. 2019;14(7):1097–9.
Kayakabe K, Kuroiwa T, Sakurai N, et al. Interleukin-1β measurement in stimulated whole blood cultures is useful to predict response to anti-TNF therapies in rheumatoid arthritis. Rheumatology (Oxford). 2012;51(9):1639–43.
Cure MC, Cure E, Kalkan Y, et al. The protective effect of adalimumab on renal injury in a model of abdominal aorta cross-clam**. Adv Clin Exp Med. 2016;25(2):219–26.
Puchalski T, Prabhakar U, Jiao Q, Berns B, Davis HM. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2010;16(5):1652–61.
Kobori H, Mori H, Masaki T, Nishiyama A. Angiotensin II blockade and renal protection. Curr Pharm Des. 2013;19(17):3033–42.
Si X, Li P, Zhang Y, Zhang Y, Lv W, Qi D. Renoprotective effects of olmesartan medoxomil on diabetic nephropathy in streptozotocin-induced diabetes in rats. Biomed Rep. 2014;2(1):24–8.
Gross O, Schulze-Lohoff E, Koepke ML, et al. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol Dial Transpl. 2004;19(7):1716–23.
Palomo M, Vera M, Martin S, et al. Up-regulation of HDACs, a harbinger of uraemic endothelial dysfunction, is prevented by defibrotide. J Cell Mol Med. 2020;24(2):1713–23.
Kshirsagar AV, Shoham DA, Bang H, Hogan SL, Simpson RJ Jr, Colindres RE. The effect of cholesterol reduction with cholestyramine on renal function. Am J Kidney Dis. 2005;46(5):812–9.
Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129(9):1022–32.
Devassy JG, Yamaguchi T, Monirujjaman M, et al. Distinct effects of dietary flax compared to fish oil, soy protein compared to casein, and sex on the renal oxylipin profile in models of polycystic kidney disease. Prostaglandins Leukot Essent Fatty Acids. 2017;123:1–13.
Thomas MC. The potential and pitfalls of GLP-1 receptor agonists for renal protection in type 2 diabetes. Diabetes Metab. 2017;43(Suppl 1):2S20–7.
Dolman ME, van Dorenmalen KM, Pieters EH, et al. Imatinib-ULS-lysozyme: a proximal tubular cell-targeted conjugate of imatinib for the treatment of renal diseases. J Control Rel. 2012;157(3):461–8.
Tang TT, Lv LL, Pan MM, et al. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018;9(3):351.
Hanna RM, Barsoum M, Arman F, Selamet U, Hasnain H, Kurtz I. Nephrotoxicity induced by intravitreal vascular endothelial growth factor inhibitors: emerging evidence. Kidney Int. 2019;96(3):572–80.
Abbas A, Mirza MM, Ganti AK, Tendulkar K. Renal toxicities of targeted therapies. Target Oncol. 2015;10(4):487–99.
Yao X, Tangri N, Gersh BJ, et al. Renal outcomes in anticoagulated patients with atrial fibrillation. J Am Coll Cardiol. 2017;70(21):2621–32.
Todd DJ, Kay J. Gadolinium-induced fibrosis. Annu Rev Med. 2016;67:273–91.
Rashid S, Ali N, Nafees S, Hasan SK, Sultana S. Mitigation of 5-Fluorouracil induced renal toxicity by chrysin via targeting oxidative stress and apoptosis in wistar rats. Food Chem Toxicol. 2014;66:185–93.
Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med. 2006;17(5):343–8.
Abd-Elsalam S, Abo-Amer YE, El-Abgeegy M, et al. Efficacy and safety of ombitasvir/paritaprevir/ritonavir/ribavirin in management of Egyptian chronic hepatitis C virus patients with chronic kidney disease: a real-life experience. Medicine (Baltimore). 2020;99(42): e21972.
Czock D, Scholle C, Rasche FM, Schaarschmidt D, Keller F. Pharmacokinetics of valganciclovir and ganciclovir in renal impairment. Clin Pharmacol Ther. 2002;72(2):142–50.
Houghton GW, Dennis MJ, Gabriel R. Pharmacokinetics of metronidazole in patients with varying degrees of renal failure. Br J Clin Pharmacol. 1985;19(2):203–9.
Rungkitwattanakul D, Chaijamorn W, Charoensareerat T, et al. Optimal levofloxacin dosing regimens in critically ill patients with acute kidney injury receiving continuous renal replacement therapy. J Crit Care. 2021;63:154–60.
Yang LJ, Hsu SM, Wu PH, et al. Association of digoxin with mortality in patients with advanced chronic kidney disease: a population-based cohort study. PLoS ONE. 2021;16(1): e0245620.
Acknowledgements
Thanks to Zhao’s team (Offical Wechat Account: SCIPhD) of Sheng **n Zhu Shou for suggestions and grammarly editing on the article.
Funding
This study was supported by the National Key R&D Program of China (Grant Number 2020YEC2005000). The founder supports the scientific research with non-profit interests and provides the researcher with remuneration.
Author information
Authors and Affiliations
Contributions
LZ and JC conceived and wrote the manuscript. JX contributed to the conception and further writing. ZY reviewed and edited this article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Cai, J., **ao, J. et al. Identification of core genes and pathways between geriatric multimorbidity and renal insufficiency: potential therapeutic agents discovered using bioinformatics analysis. BMC Med Genomics 15, 212 (2022). https://doi.org/10.1186/s12920-022-01370-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01370-1