
Abstract
Background
Leber’s congenital amaurosis (LCA) is a severe hereditary retinopathy disease that is characterized by early and severe reduction of vision, nystagmus, and sluggish or absent pupillary responses. To date, the pathogenesis of LCA remains unclear, and the majority of cases are caused by autosomal recessive inheritance. In this study, we explored the variant in the Crumbs homologue 1 (CRB1) gene in a Chinese family with LCA.
Methods
We conducted comprehensive ocular examinations and collected 5 ml of blood samples from members of a Chinese family with LCA. A pathogenic variant was identified by capturing (the panel in NGS) and Sanger sequencing validation.
Results
A nonsense variant (c.1499C>G) in the 6th exon of CRB1 gene in a Chinese family with LCA was identified, which predicted a change in the protein p. S500*, may lead to loss of gene function. We summarized the 76 variants reported thus far in CRB1 that caused LCA8.
Conclusions
This study reported a novel variant c.1499C>G (p. S500*) of the CRB1 gene occurred in a Chinese family with LCA, thus expanding the spectrum of CRB1 variants causing LCA.
Similar content being viewed by others

Introduction
Since Theodore Leber first described Leber’s congenital amaurosis (LCA) in 1869, a great deal of information about LCA has been revealed, including both clinical characteristics and molecular genetics. LCA, a rare but important juvenile retinal dystrophy, is an inherited retinal disorder most often diagnosed in infancy in the first 6 months of life and characterized by the presence of nystagmus, poor visual acuity (VA), and a severely reduced or nondetectable electroretinogram [1, 2]. The global incidence of LCA ranges from 1/81,000 to 1/30,000 among newborn babies. Although the incidence is low, this disorder also causes blindness in 20% of LCA school-age children and accounts for approximately 5% of all cases of hereditary retinopathy [3, 4]. LCA is currently categorized into 21 types according to the pathogenic genes, with autosomal recessive inheritance as the dominant. LCA8 is caused by a homozygous or compound heterozygous variant in the CRB1 gene (OMIM *604210) on chromosome 1q31.
Material and methods
The proband (Fig. 1), a 2-year-old girl, her parents complained that she has poor eyesight in both eyes and could not accurately grasp objects. She was unable to comply with the detailed eye examination. Under the guidance of the paediatrician, the opportunity for examination was obtained through oral anaesthesia. Her parents and sister underwent detailed eye examinations, including binocular corrected visual acuity, slit lamp examination, fundus photography, macular and optic disc OCT scanning, and electroretinogram (ERG).

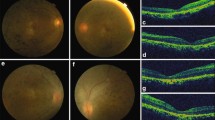
Pedigree of the LCA family with a CRB1 variant, sequencing chromatogram, and diagnostic fundus. a Pedigree of the LCA family with a CRB1 variant. The proband is marked by an arrow, black symbols denote affected members, white symbols denote unaffected members, squares denote males, and circles denote females. b Sequencing chromatograms. The affected proband showed a homozygous variant in the CRB1 gene: nucleotide 1499 changed from a cytosine to guanine (c.1499C>G) homozygous variant, resulting in a nonsense variant of amino acids (p. S500*), her parents and sister show a heterozygous variant at the same site. c Diagnosis of the fundus. Both eyes of the proband showed pigmentation of the retina at the posterior pole that was peppery and salt-like, and the macular area was a mass of lesions with a large amount of pigmentation. d Her 5-year-old sister’s fundus is normal, and the same is true for her parents
Five millilitres of peripheral blood was obtained from each of the 4 subjects (II5, II8, III1, and III2) and collected in EDTA tubes for DNA extraction. The panel (463 genes related to ocular diseases) in next-generation sequencing (NGS) was used to capture the target gene. Then, Sanger sequencing was performed to validate the variants from 22 candidate genes. The transcript used to identify the variant in the CRB1 gene was NM_201253.
Results
The proband's parents came from two unrelated families, with no consanguineous or inbreeding. Except for the proband, neither parent had a family member with similar eye disease (Fig. 1a).
After panel capture, 22 candidate genes remained. Primers were designed for each candidate gene, PCR was performed, and then, first-generation Sanger sequencing was used to verify the target gene. Finally, we obtained the target co-isolation gene in this family. Sequencing chromatograms: the proband shows a homozygous variant in the CRB1 gene, nucleotide 1499 changed from cytosine (C) to guanine (G) c.1499C>G(p. S500*), her parents and sister showed a heterozygous variant at the same site (Fig. 1b).
The proband's eyes showed horizontal pendulum nystagmus. On examination, her eyes were in a normal position, the cornea and lens were clear, fundoscopy showed that the colour of the optic disc in both eyes was light, and the blood vessels from both eyes were thin and narrow (Fig. 1c). The pigmentation of the retina at the posterior pole was peppery and salt-like, and the macular area was a mass of lesions with much pigmentation.
After detailed eye examinations, the proband’s parents and sister showed normal results (Fig. 1d; Table 1).
For LCA, the criteria include signs of blindness or severe visual impairment from birth or within the first year of life, an ERG reduction of more than 50%, and congenital nystagmus [5]. Fundus examinations could reveal diagnostic clues, including peripheral pigmentary retinopathy, central maculopathy with or without bull’s eye pattern, or even macular atrophy. In addition, indispensable, molecular confirmation is needed.
In our study, the proband's eye examinations and genetic tests were consistent with the diagnosis of LCA. The homozygous variant in the 6th exon of CRB1: nucleotide 1499 changed from cytosine to guanine (c.1499C>G), resulting in a nonsense variant of amino acids (p. S500*), which has not been reported before. The proband's parents and sister had heterozygous variation at this site. According to the ACMG (American College of Medical Genetics and Genomics) guidelines, the variant was preliminarily determined to be pathogenic: PVS1 + PM2 + PM3_Supporting (hom). PVS1: This variant is a zero-effect variant (nonsense variant), which may lead to loss of gene function; PM2: This variant frequency in the database of the normal population (1000g2015aug_all, ESP6500si, GnomAD_Genome_ALL, GnomAD_Genome_EAS, etc.) is “-”, which means the variant was not detected; PM3_Supporting (HOM): This variant is a homozygous rare variant. No correlation of this locus was reported in the literature database. No pathogenicity analysis results were found in the ClinVar database. Our study expands the spectrum of CRB1 variants causing LCA.
We used the ScanProsite tool (https://prosite.expasy.org/scanprosite/) to examine the secondary structure of the CRB1 protein. The nonsense variant c.1499C>G (p. S500*) is in the laminin G domain profile 485–670: score = 32.931.L, Yang, et al. also reported a nonsense variant c.1576C>T(p. R526X) in this domain [6]. Laminin G is an approximately 180 amino acid long domain found in a large and diverse set of extracellular proteins. It often occurs in multiple copies, probably serving as general protein interaction domains that bind target proteins and other macromolecules, such as carbohydrates. In most proteins, the precise function of the laminin G domain is unknown. A large number of ligands in the G domain of laminin have been reported, including heparin, sulfatides, integrins, dystroglycan, nidogen, and fibulin. In neurexin, the G domain is known to bind neurexophilins, α-latrotoxin and neuroligins [7, 8].
Another anatomical feature of LCA includes decreased thickness in different layers, especially in the outer nuclear layer (ONL), loss of integrity in the ellipsoid zone, and disorganized macular atrophy [9]. Unfortunately, the proband we reported was too young to cooperate with optical coherence tomography (OCT) and ERG examination, so we could not analyse the clinical features of these two aspects.
LCA caused by CRB1
In 2004, Hanein et al. [10] reported a comprehensive mutational analysis of all known genes in 179 unrelated LCA patients, including 52 familial and 127 sporadic cases. The results showed that variants were identified in 47.5% of patients. GUCY2D appeared to account for most LCA cases in their series (21.2%), followed by CRB1 (10%), RPE65 (6.1%), RPGRIP1 (4.5%), AIPL1 (3.4%), TULP1 (1.7%), and CRX (0.6%). Three years later, Francesca Simonelli et al. [11] analysed 95 patients in Italy with LCA. They identified some novel variants that occurred frequently in the RPE65 (8.4%), CRB1 (7.4%), and GUCY2D (5.2%) genes. Through a detailed ophthalmic evaluation of patients with the variant, they found that CRB1 variants were associated with reduced retinal thickness and a coarsely laminated retina (by OCT). In London, Henderson, R.H., et al. acquired DNA samples from 250 probands with LCA/early childhood-onset retinal dystrophy (EORD). They analysed using the LCA chip, and CRB1 variants were identified in 21 probands [12]. Corton et al. enrolled 404 Spanish patients in the study, 114 of which suffered from LCA and 290 from EORP (early-onset RP). Their study revealed that 11% of Spanish patients carried variants in CRB1, ranging from 9% of EORP to 14% of LCA cases. More than three-quarters of the variants identified were first described in their study [13].
Li** Yang et al. [6] used 18 cases presenting with LCA to identify disease-causing variants. They reported compound heterozygous variants of the CRB1 gene, which included three novel heterozygous variants: c.3059delT (p. M1020Sfs*1), c.3460T>A (p. C1154S), and c.4207G>C (p.E1403Q). Hosono et al. reported variants of LCA- and inherited retinal dystrophy (IRD)-associated genes in 34 Japanese families, which is the first study to conduct a next-generation sequencing (NGS)-based molecular diagnosis of a large Japanese LCA cohort and achieved a detection rate of approximately 56%. Their results showed that the most frequently mutated genes were CRB1, NMNAT1, and RPGRIP1 [14]. Recently, Zhu et al. [15] enrolled 37 patients with strictly defined LCA in a cohort of IRD in ten years (2009–2019). Their results revealed that the CRB1 gene occupied a greater proportion (27%) of associated LCAs in the western Chinese population.
CRB1 variants are a common cause of LCA, and related variants include substitution, deletion, duplication and insertion. Table 2 lists the variants in LCA caused by CRB1, which include variant types, sites, corresponding amino acid changes and regions in recent years. These results are for readers' verification and reference.
“–”: not applicable
To date, a total of 76 CRB1 variants have caused LCA. Furthermore, it has been reported that variants in CRB1 are responsible for 7.4%-27% of LCA in different populations. The pathogenic variants were mainly substitution and deletion, including duplication, insertion, which reflected the richness of variant types (Table 3). The variant sites of LCA8 were mainly concentrated in exons 6, 7 and 9 of CRB1, and clear pathogenic sites were found in all exons except exons 4, 5 and 10, indicating the universality of variant regions (Table 4). The reported cases involved more than 10 countries and regions, including China, England, Japan, Spain and Italy, which also showed that the global coverage of LCA caused by CRB1 is extensive.
Other diseases of retinal dystrophy caused by CRB1 variants
In addition to LCA, variants in CRB1 are associated with several other diseases of retinal dystrophy: Rosa Riveiro-Alvarez, et al. [27] reported an early-onset RP phenotype in a Spanish family caused by the Nonsense CRB1 c.2843G>A(p. C948Y) variant. Two CRB1 substitution variants, c.3991C>T(p. R1331C) and c.4142C > T(p. P1381 L), were reported to illustrate a novel presentation of macular dystrophy caused by CRB1 variants by Stephen H. Tsang et al. [28]. Arif O. Khan et al. uncovered a homozygous CRB1 variant c.80G>T(p. C27F) in three siblings with childhood cone-rod dystrophy and macular cystic degeneration in a family [29]. Ajoy Vincent et al. reported biallelic variants (p. G123C and p. C948Y, p. I167_G169del and p. R764C) in CRB1 in two families caused autosomal recessive familial foveal retinoschisis, which may be the mildest end of the spectrum of CRB1-related diseases [30]. Benjamin K. Ghiam et al. reported that a novel variant c.4014T>A in CRB1 was related to retinal degeneration and may portend a poor prognosis for CME responsiveness to therapy [31].
Discussion
LCA is the earliest and most severe hereditary retinopathy, in which the function of cone-rod cells in both eyes is completely lost at birth or within one year after birth, leading to congenital blindness in infants. The majority of cases are caused by autosomal recessive inheritance. Typical characteristics of LCA include early and severe reduction of vision associated with nystagmus, photophobia, sluggish or absent pupillary responses, finger pressure on eyeballs, fundus appearance, ranging from normal, maculopathy, to typical RP-like abnormalities, and electroretinogram showed that A and B waves were flat and even severely reduced to nondetectable. It can also be accompanied by keratoconus, hyperopia, developmental delay and nervous system abnormalities [32].
In some cases/reports, there are many similar clinical features between LCA and early-onset RP, and even the diagnosis is ambiguous[33]. Early-onset RP is usually considered to be a relatively milder form, in which patients do not have a congenital onset of visual impairment. We could distinguish the following phenotypes: LCA, early onset retinal degeneration; RP, presence of preservation of the para-arteriolar retinal pigment epithelium and Coats-like vasculopathy[34].
To date, 28 genes involved in the pathogenesis of LCA [35]. CRB1 belongs to LCA8. The CRB1 gene maps to chromosome 1q31.3 and is composed of 12 exons; the longest isoform consists of 1,406 amino acids. This gene encodes a protein that is similar to the Drosophila crumb protein and localizes to the inner segment of mammalian photoreceptors. In Drosophila, crumbs localize to the stalk of the fly photoreceptor and may be a component of the molecular scaffold that controls proper development of polarity in the eye[36], and CRB1 has been found to be important in maintaining cellular polarity[37].
In the mouse retina, CRB1 is expressed in the inner segment of the photoreceptors and Muller cells to maintain adequate morphogenesis and polarity in retinal development[38]. Therefore, CRB1 gene variants often lead to a variety of retinal dystopathies, including retinitis pigmentosa (RP), LCA, and macular dystrophy. Approximately 9–17% of LCA cases have been related to CRB1 variants, which is especially higher in the Chinese population[39, 40]. A wide variety of visual acuity was noted in patients with variants in CRB1, ranging from 20/30 to NLP[10, 41].
Among LCA, RPE65 variants were almost always associated with normal macular thickness, as assessed by OCT, whereas CRB1 variants were associated with reduced retinal thickness and a coarsely laminated retina. Fundus abnormalities were more heterogeneous in carriers of CRB1 variants. In fact, some scholars observed salt-and-pepper retinal dystrophy in younger patients and subsequently massive spicular and not nummular pigmentation at the posterior pole, which was reported to be a phenotypic feature of carriers of CRB1 variants[11]. Saloni Walia et al. [42]. Through a multicentre retrospective observational study with 169 patients with LCA, variants in RPE65 (LCA-Type Ii) and CRB1 (LCA-8) may be associated with a relatively better VA in early life compared with other gene variants. The onset of the symptoms of LCA after the age of 1 year is also associated with an overall better VA prognosis.
Conclusions
LCA is one of the earliest and most severe forms of inherited IRD. Patients suffer from severe visual impairment during childhood, with their vision continuously deteriorating, the final outcome of which is usually complete loss of vision by their thirties or forties[43]. Therefore, it is very important to find an effective treatment. Albert et al. provided an entirely new dimension in ocular therapeutics for gene therapy to LCA2. Patients with LCA2 who received AAV2.hRPE65v2 by subretinal injection showed evidence of improvement in retinal function, pupillary light reflex, and reduction in nystagmus. These clinical trials are approaches to the treatment of LCA and possibly other forms of retinal degeneration[44].
However, much is still unknown about the pathogenesis of LCA. With the improvement of next-generation sequencing technology and the application of various molecular biological means, research on corresponding cell functions, the identification of gene subtypes and the establishment of animal models have greatly promoted our understanding of LCA. These latest advances provide a steady stream of evidence for a better understanding and treatment of LCA in the future. These findings may be useful for faster gene diagnosis, prenatal testing, the development of potential gene therapies, and for improving the understanding of the molecular pathogenesis of LCA.
Availability of data and materials
The relevant data were generated during this study and included in this article. Raw sequence data were not available in this article, as no datasets were generated during the current study. The corresponding author Liwei Zhang (drzhangliwei@163.com) should be contacted if someone wants to request the data from this study.
References
Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. 2004;49(4):379–98.
Traboulsi EI, Koenekoop R, Stone EM. Lumpers or splitters? The role of molecular diagnosis in Leber congenital amaurosis. Ophthalmic Genet. 2006;27(4):113–5.
Koenekoop RK, Lopez I, den Hollander AI, Allikmets R, Cremers FP. Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions. Clin Exp Ophthalmol. 2007;35(5):473–85.
Chen TC, Huang DS, Lin CW, Yang CH, Yang CM, Wang VY, Lin JW, Luo AC, Hu FR, Chen PL. Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan. NPJ Genom Med. 2021;6(1):16.
Booij JC, Florijn RJ, ten Brink JB, Loves W, Meire F, van Schooneveld MJ, de Jong PT, Bergen AA. Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med Genet. 2005;42(11): e67.
Li** Yang ea. Novel mutations of CRB1 in Chinese families presenting with retinal dystrophies. Mol Vis. 2014;20:359–67.
Sung U, et al. Localization of heparin binding activity in recombinant laminin G domain. EurJBiochem. 1997;250(1):138–43.
Beckmann G, et al. Merging extracellular domains: fold prediction for laminin g-like and amino-terminal thrombospondin like modules based on homology to pentraxins. J Mol Biol. 1998;275(5):725–30.
Cideciyan AV, Jacobson SG. Leber congenital amaurosis (LCA): potential for improvement of vision. Investig Ophthalmol Vis Sci. 2019;60(5):1680–95.
Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23(4):306–17.
Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, Fossarello M, Signorini S, Bertone C, Galantuomo S, et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci. 2007;48(9):4284–90.
Henderson RH, Mackay DS, Li Z, Moradi P, Sergouniotis P, Russell-Eggitt I, Thompson DA, Robson AG, Holder GE, Webster AR, et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol. 2011;95(6):811–7.
Corton M, et al. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet J Rare Dis. 2013;8:20.
Hosono K, Nishina S, Yokoi T, Katagiri S, Saitsu H, Kurata K, Miyamichi D, Hikoya A, Mizobuchi K, Nakano T, et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci Rep. 2018;8(1):8279.
Zhu L, Ouyang W, Zhang M, Wang H, Li S, Meng X, Yin ZQ. Molecular genetics with clinical characteristics of Leber congenital amaurosis in the Han population of western China. Ophthalmic Genet. 2021;42(4):392–401.
McKibbin M, et al. Genotype-phenotype correlation for leber congenital amaurosis in northern pakistan. Arch Ophthalmol. 2010;128(1):107–13.
Beryozkin A. Mutations in CRB1 are a relatively common cause of autosomal recessive early-onset retinal degeneration in the Israeli and Palestinian populations. Investig Ophthalmol Vis Sci. 2013;54:2068–75.
Ahmed Khan S, Richard Nestel A. CRB1 gene mutation causing different phenotypes of leber congenital amaurosis in siblings. J Ophthalmic Vis Res. 2019;14(4):518–24.
Li L, **ao X, Li S, Jia X, Wang P, Guo X, Jiao X, Zhang Q, Hejtmancik JF. Detection of variants in 15 genes in 87 unrelated Chinese patients with Leber congenital amaurosis. PLoS ONE. 2011;6(5):e19458.
Seong M-W. Molecular characterization of Leber congenital amaurosis in Koreans. Mol Vis. 2008;14:1429–36.
Chen Y, Zhang Q, Shen T, **ao X, Li S, Guan L, Zhang J, Zhu Z, Yin Y, Wang P, et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Investig Ophthalmol Vis Sci. 2013;54(6):4351–7.
Mea S. CRB1-related leber congenital amaurosis: reporting novel pathogenic variants and a brief review on mutations spectrum. Iran Biomed J. 2019;23(5):362–8.
Vamos R, Kulm M, Szabo V, Ahman A, Lesch B, Schneider M, Varsanyi B, Nagy ZZ, Nemeth J, Farkas A. Leber congenital amaurosis: first genotyped Hungarian patients and report of 2 novel mutations in the CRB1 and CEP290 genes. Eur J Ophthalmol. 2016;26(1):78–84.
Lin Li ea. Lack of phenotypic effect of triallelic variation in SPATA7 in a family with Leber congenital amaurosis resulting from CRB1 mutations. Mol Vis. 2011;17:3326–32.
Skorczyk-Werner A, Niedziela Z, Stopa M, Krawczynski MR. Novel gene variants in Polish patients with Leber congenital amaurosis (LCA). Orphanet J Rare Dis. 2020;15(1):345.
Hollander AI. Leber congenital amaurosis and retinitis pigmentosa with coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203.
Riveiro-Alvarez R. Molecular analysis of ABCA4 and CRB1 genes in a Spanish family segregating both Stargardt disease and autosomal recessive retinitis pigmentosa. Mol Vis. 2008;14:262–7.
Tsang SH, Burke T, Oll M, Yzer S, Lee W, **e YA, Allikmets R. Whole exome sequencing identifies CRB1 defect in an unusual maculopathy phenotype. Ophthalmology. 2014;121(9):1773–82.
Khan AO, Aldahmesh MA, Abu-Safieh L, Alkuraya FS. Childhood cone-rod dystrophy with macular cystic degeneration from recessive CRB1 mutation. Ophthalmic Genet. 2014;35(3):130–7.
Vincent A, Ng J, Gerth-Kahlert C, Tavares E, Maynes JT, Wright T, Tiwari A, Tumber A, Li S, Hanson JV, et al. Biallelic mutations in CRB1 underlie autosomal recessive familial foveal retinoschisis. Investig Ophthalmol Vis Sci. 2016;57(6):2637–46.
Ghiam BK, Wood EH, Thanos A, Randhawa S. CRB1 related retinal degeneration with novel mutation. Am J Ophthalmol Case Rep. 2020;18:100699.
den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419.
Jalkh N, Guissart C, Chouery E, Yammine T, El Ali N, Farah HA, Megarbane A. Report of a novel mutation in CRB1 in a Lebanese family presenting retinal dystrophy. Ophthalmic Genet. 2014;35(1):57–62.
Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, Leveillard T, Letexier M, Saraiva JP, Lonjou C, et al. CRB1 mutations in inherited retinal dystrophies. Hum Mutat. 2012;33(2):306–15.
Kondkar AAA-AK. Leber congenital amaurosis: current genetic basis, scope for genetic testing and personalized medicine. Exp Eye Res. 2019;189: 107834.
Izaddoost S. Drosophila Crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature. 2002;416:178–83.
Tepass U. Crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelia cells and required for organization of epithelia. Cell. 1990;61:787–99.
Alves CH, Pellissier LP, Wijnholds J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog Retin Eye Res. 2014;40:35–52.
Wang H, Wang X, Zou X, Xu S, Li H, Soens ZT, Wang K, Li Y, Dong F, Chen R, et al. Comprehensive molecular diagnosis of a large chinese leber congenital amaurosis cohort. Invest Ophthalmol Vis Sci. 2015;56(6):3642–55.
Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–54.
Jea LAJ. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. 2001;119:415–20.
Walia S, Fishman GA, Jacobson SG, Aleman TS, Koenekoop RK, Traboulsi EI, Weleber RG, Pennesi ME, Heon E, Drack A, et al. Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology. 2010;117(6):1190–8.
Sharif W, Sharif Z. Leber’s congenital amaurosis and the role of gene therapy in congenital retinal disorders. Int J Ophthalmol. 2017;10(3):480–4.
Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–8.
Acknowledgements
The authors thank the patients and all family members for their participation in this study. Dr. Min Hu (fudanhumin123@sina.com) and Dr. Liwei Zhang (drzhangliwei@163.com) are co-corresponding authors for this paper.
Funding
This study was supported by the National Natural Science Foundation of China (81860171), Medical Reserve Talents of Yunnan Province (H-2018020),Leading the charge of Yunnan Province Health System (L-2018018),and High-level Talent Training Support Program Special Famous Doctors of Yunnan Province (YNWR-MY-2018-006).
Author information
Authors and Affiliations
Contributions
WD and TZ carried out the experiments and drafted the manuscript. HJ and MZ prepared the figures and tables. LZ and MH designed and funded this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
This study was performed in agreement with the Declaration of Helsinki. It was reviewed by the research unit's professional ethics committee, and informed consent was obtained and signed by the investigator. All experimental protocols were approved by the Affiliated Hospital of Yunnan University, and methods were carried out in accordance with relevant guidelines and regulations, including any relevant details. Informed written consent was taken from all participants and legal guardians of children.
Consent for publication
Written informed consent was obtained from the guardians (parents) of the patients, and they consented to the publication of the study. The guardian (parent) of the patients consented for their medical information to be published.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Duan, W., Zhou, T., Jiang, H. et al. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics 15, 197 (2022). https://doi.org/10.1186/s12920-022-01356-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01356-z