
Abstract
Background
Rice grain size (GS) is an essential agronomic trait. Though several genes and miRNA modules influencing GS are known and seed development transcriptomes analyzed, a comprehensive compendium connecting all possible players is lacking. This study utilizes two contrasting GS indica rice genotypes (small-grained SN and large-grained LGR). Rice seed development involves five stages (S1–S5). Comparative transcriptome and miRNome atlases, substantiated with morphological and cytological studies, from S1–S5 stages and flag leaf have been analyzed to identify GS proponents.
Results
Histology shows prolonged endosperm development and cell enlargement in LGR. Stand-alone and comparative RNAseq analyses manifest S3 (5–10 days after pollination) stage as crucial for GS enhancement, coherently with cell cycle, endoreduplication, and programmed cell death participating genes. Seed storage protein and carbohydrate accumulation, cytologically and by RNAseq, is shown to be delayed in LGR. Fourteen transcription factor families influence GS. Pathway genes for four phytohormones display opposite patterns of higher expression. A total of 186 genes generated from the transcriptome analyses are located within GS trait-related QTLs deciphered by a cross between SN and LGR. Fourteen miRNA families express specifically in SN or LGR seeds. Eight miRNA-target modules display contrasting expressions amongst SN and LGR, while 26 (SN) and 43 (LGR) modules are differentially expressed in all stages.
Conclusions
Integration of all analyses concludes in a “Domino effect” model for GS regulation highlighting chronology and fruition of each event. This study delineates the essence of GS regulation, providing scope for future exploits. The rice grain development database (RGDD) ( www.nipgr.ac.in/RGDD/index.php; https://doi.org/10.5281/zenodo.7762870) has been developed for easy access of data generated in this paper.
Similar content being viewed by others


Background
Rice is the primary food source for over 3.5 billion people [7] or cell size [8] or both [9]. This increases sink area for acquiring storage compounds, leading to increased grain weight, thus, establishing a positive correlation between seed size and grain yield [10]. The quest for understanding GS regulation in rice is challenging. As a quantitative trait, it undergoes polygenic regulation and is under environmental influence. Furthermore, various traits of rice grain, namely, length, width, and weight are associated with each other and one gene/quantitative trait locus (QTL) may affect more than one such trait [11].
Grain development in rice generally occurs over a month and can be distinguished by three land mark events viz., cell division, initiation of organs, and maturation [12, 13]. The first land mark event extends from 0 to 2 days after pollination (DAP) where extensive cell division occurs immediately after anthesis followed by triple fusion to form a middle-globular embryo and a syncytial endosperm. Organ initiation on the embryo and cellularization of endosperm begin by 3–4 DAP and are marked as the second landmark process. Maturation involves organ enlargement and maturation of embryo and endosperm, including grain filling, and extends from 5 to 29 DAP. Further, maturation can be divided into distinct sub-stages which include individual organ enlargement on embryo and endoreduplication of endosperm (5–10 DAP), embryo maturation and programmed cell death (PCD) in endosperm (11–20 DAP), and dormant embryo and dehydration of endosperm (21–29 DAP). Storage accumulation occurs during maturation phase. Storage is related to grain filling, which is majorly contributed by carbohydrate and seed storage protein accumulation. Altogether, the process of rice seed development has been defined by five stages as S1 (0–2 DAP), S2 (3–4 DAP), S3 (5–10 DAP), S4 (11–20 DAP), and S5 (21–29 DAP) [12, 14].
GS regulation in rice is extremely diverse with involvement of genes controlling hormones [15], G-protein signaling [33], stress tolerance [34, 35], grain quality, and hybrid vigor [2: Table S10). All these detected QTLs were mapped on 12 chromosomes explaining 1.5–30% phenotypic variation for grain size/weight with a significant logarithm of the odds score (LOD) (2.5–13.0) in rice. Further, genes from each of the abovementioned transcriptome analysis identified to delineate pathways controlling rice GS differentiation, which were either stage-specific or were highly differentially regulated between SN and LGR (Fig. 9, Additional file 2: Tables S5, S6, S7, S8 and S9), were overlayed with the grain size/weight QTLs. A total of 186 of these genes (Additional file 2: Table S11) were found to be located within QTL genomic intervals. These were contributed by 31 genes which were commonly differentially regulated in a stage-specific manner in SN and LGR, 15 cell cycle-related genes which were specifically upregulated in either SN S5 or LGR S3 stage, 38 LGR S3 preferential genes, 45 TFs belonging to families with stage-preferential expression in a stage of either SN or LGR, 50 phytohormone related genes which had a different pattern and level of expression between SN and LGR, and seven genes with opposite regulation in SS or CS comparisons whose upstream miRNAs also show opposite regulation. These 186 genes (including regulatory genes) present in the QTL genomic regions associated with GS traits give a strong indication of their roles in the process, and thus should be explored especially as promising candidate genes by their detailed functional characterization in rice.
Novel miRNA-target modules in rice GS regulation
In rice seeds, miRNAs are known to function both in early seed development [57] and grain filling [58]. Using exactly the same seed development tissues (S1–S5) and flag leaf, as for transcriptome, miRNA expression profiles were generated for SN and LGR. Small RNA sequencing generated a total of 14 Gb clean data for 36 samples (Additional file 2: Table S12) with average Pearson’s correlation of 0.85 between biological replicates (Additional file 2: Table S13). miRNAs with TPM ≥ 50 [59, 60] were counted as expressed. The data was validated by stem-loop qRT-PCR of miR530-5p (Additional file 1: Figure S12A). A total of 193 (SN) and 196 (LGR) miRNAs were expressed, belonging to 70 and 76 families, of which four and 10 families were specific to SN and LGR, respectively (Additional file 1: Figure S12B, Additional file 2: Table S14). The numbers of total and specific miRNAs expressed in each stage were more in LGR (Additional file 1: Figure S12C). This was in conjunction with lesser number of expressed genes in LGR (Additional file 1: Figure S3). LGR also had a greater number of differentially expressed miRNAs (DEMs) than SN (Additional file 1: Figure S13A). Comparison of DEMs amongst CS and same SS stages of SN and LGR showed that most miRNAs exhibited similar regulation between SN and LGR in both comparisons (Additional file 1: Figure S13B, Additional file 2: Table S15).
Further, miRNAs and their targets with negative correlation in expression patterns were identified. DEMs present throughout seed development in SN or LGR and their targets were delineated to extract miRNA-target modules pertinent to GS (Additional file 1: Figure S14A, B, Additional file 2: Table S16). Thirteen miRNAs which were upregulated in all the five stages in SN had 85 targets which were downregulated in all stages in SN (Additional file 1: Figure S14A). These included osa-miR1848, osa-miR319a-3p, osa-miR535-3p, osa-miR1872, osa-miR1874, and osa-miR5504, which have been discussed later. Conversely, in SN itself, 13 miRNAs which were downregulated in all the five stages had 41 targets with negatively correlated expression patterns (Additional file 1: Figure S14B, Additional file 2: Table S16). This set included osa-miR1432 and osa-miR397, which have been discussed. Targets of these 13 miRNAs belonged to functional categories including cell wall synthesis, amino acid metabolism, TFs, protein post-translational modification, signaling, and transport. In LGR, 21 miRNAs that were upregulated in all the five stages had 61 negatively correlated targets (Additional file 1: Figure S14A, Additional file 2: Table S16). Majority of these were non-conserved miRNAs (specific to rice). These included osa-miR529 and osa-miR396, which have been discussed. Targets of these 21 miRNAs belonged to the functional categories including amino acid metabolism, hormone metabolism, secondary metabolism, TFs, stress response, signaling, and transport. Twenty-two miRNAs were downregulated in all the five stages of LGR and had 38 targets (Additional file 1: Figure S14B, Additional file 2: Table S16). In this set, osa-miR166, osa-miR398, osa-miR159, osa-miR167i-3p, and osa-miR172a have been discussed. Targets of these 22 miRNAs belonged to several functional categories relevant to GS, including cell cycle, cell division, cell wall modification, cell organization, DNA synthesis, fermentation, and starch synthesis.
Five miRNAs were upregulated throughout seed development in both SN and LGR (Fig. 8A). They had a total of nine targets which showed negative correlation in expression pattern and were downregulated throughout seed development in both SN and LGR. Similarly, five miRNAs were downregulated throughout seed development in both SN and LGR (Fig. 8B), and targeted eight genes having negative correlation in expression. These targets belonged to functional categories which included TFs, hormone-related genes, signaling and transport, amino acid metabolism, and light reaction. These included osa-miR396, osa-miR408-3p, osa-miR408-5p, osa-miR444, and osa-miR528, which have been detailed further on. Of these, osa-miR319 and osa-miR528-5p had multiple novel predicted targets.

Expression analysis of miRNAs-targets pairs showing negative correlation in all five seed developmental stages of both SN and LGR. A Graphs showing differential expression levels (log2fc) of five miRNAs upregulated and their targets downregulated in all five stages of SN and LGR. B Graphs showing differential expression levels (log2fc) of five miRNAs downregulated and their targets upregulated in all five stages of SN and LGR. In each graph, dotted and solid lines represent miRNAs and their targets, respectively. Names of miRNAs and their targets have been mentioned in the legends in each graph
DEMs with opposite regulation between SN and LGR should also be important players in GS (Additional file 1: Figure S13B). In terms of SS, a total of 22 miRNAs were upregulated in any stage in LGR and downregulated in the same stage in SN. Similarly, five miRNAs were upregulated in any stage in SN and downregulated in the same stage in LGR. In terms of CS, nine miRNAs were upregulated in LGR and downregulated in a comparable stage in SN, while six miRNAs were regulated in a vice versa manner. Out of these, for eight miRNAs that were upregulated in LGR and downregulated in SN, nine targets showed negative correlation in expression (Fig. 9). A prominent family which was highlighted by this analysis was osa-miR2118, which has been detailed later. No targets were found showing negative correlation for miRNAs upregulated in SN and downregulated in LGR. The targets for eight miRNAs upregulated in LGR and downregulated in SN included two genes each for lipid metabolism genes and transport, and one gene each related to cytochrome P450, UDP glucosyl and glucoronyl transferases (UGTs), and biotic stress. Therefore, these miRNA-target modules exhibiting opposite behavior amongst LGR and SN might be essential regulators of GS in rice and should be examined further.

miRNA-target modules showing opposite regulation pattern in seed developmental stages of SN and LGR. Graphs showing differential expression levels (log2fc) of eight miRNAs upregulated in same stage (i.e., SN S1-LGR S1, SN S2–LGR S2) or comparable stage (i.e., SN S1-LGR S2, SN S2-LGR S3, SN S3-LGR S4, SN S4-LGR S5) of LGR and downregulated in same or comparable stage of SN, and their targets that show negative correlation in SN and LGR (downregulated in LGR and upregulated in SN in the same stage or comparable stage). IDs of miRNA and targets have been mentioned in the legends in each graph. Dotted blue line and solid lines represent expression of miRNA and targets, respectively. In each graph, negative correlation obtained between miRNA and its target in same stages and comparable stage has been marked with purple and blue bars, respectively
Rice grain development database (RGDD) allows for easy data access
To make the transcriptome and miRNome data searchable, RGDD (www.nipgr.ac.in/RGDD/index.php) has been developed. In the transcriptome tab, Locus ID from RGAP (Rice Genome Annotation Project) can be directly entered in option 1. In case the Locus ID is not known, and the user wants to search using gene functions, then option 2 should be used. In this case, search can be made using options of the gene encoding for a TF or related to a phytohormone. Transposable elements can also be selected for or eliminated using option 2. The output shows both expression (average FPKM) and differential expression (Log2 fold change) values for all stages of both SN and LGR. The output also mentions if the gene is seed-specific or not (Additional file 1: Figure S16A). In the miRNome tab, option 1 allows for entering of a known miRNA ID. Option 2 allows to select an miRNA from a dropdown list. The output displays both the expression (average TPM) and differential expression (log2 fold change) values (Additional file 1: Figure S16B).
Discussion
Contrasting genotypes, SN and LGR
In the present study, two rice genotypes contrasting for GS have been considered for transcriptome and miRNome analyses, throughout five seed development stages, with the aim to elucidate genes and pathways contributing to increase in GS. In such extensive studies, there is a possibility of data dilution by genes reflecting genotypic differences. In order to minimize this, flag leaf for both genotypes has been used as a control to calculate DEGs involved in seed development, individually for SN and LGR. Subsequently, these DEGs have been compared amongst SN and LGR to elucidate the ones with different expression patterns and levels. Here, flag leaf has served as a control to remove genes which might have expressed at high levels in both leaf and seed in a particular genotype only. If not eliminated by this method, these genes would have been highlighted as DEGs and would be due to genotypic differences, and not represent an actual role in GS. The comparative methodology used in the present study has often been used to examine two plant varieties contrasting for a given trait. Temporal transcriptome analysis for cold tolerance, in two contrasting cultivars of tobacco, Tai tobacco (TT, cold susceptibility) and Yan tobacco (YT, cold resistance), has been used to identify DEGs in both cultivars after comparing with the corresponding control (without cold treatment) for each cultivar [61]. Comparative transcriptomics study in the root and shoot tissues of N-efficient (PBW677) and N-inefficient (703) cultivars of wheat have revealed the genes that regulate nitrogen use efficiency [62]. PBW677 has considerably more abundant DEGs compared to PBW703. Two contrasting peanut cultivars, Zhonghuahei 1 and Zhongkaihua 151, with high and low free amino acids in mature seeds, respectively, have been compared by metabolomics and transcriptomic approaches to identify the regulatory network of amino acid metabolism [63]. Recently, a study integrated metabolome with transcriptome analyses in order to have a better understanding of the metabolite profiles and molecular mechanisms regulating different cane traits, namely, brix, rind color, and textures in the stems and leaves of contrasting sugarcane varieties FN41 and 165402 [64]. Two contrasting teak cultivars, T. grandis “** rice grain transcriptome using the agilent microarray platform. Methods Mol Biol. 2019;1892:277–300." href="/article/10.1186/s12915-023-01577-3#ref-CR67" id="ref-link-section-d343189e2129">67]. Transcriptome of whole rice grains has been used to study grain filling under soil drying conditions [68]. Entire panicles have been used for studying transcriptome rhythms due to warm night temperature [69]. Transcriptome analyses have been performed on mutant and wild-type 12 DAP whole grains to understand role of Fernonia-like receptor genes in GS [70]. Complete caryopsis has been used to examine the transcriptome and phytohormone changes contributing to grain chalkiness [57, 81]. miR1874 was upregulated throughout seed development in both SN and LGR (Fig. 8A). It specifically expresses in develo** seeds of both japonica cultivar Nipponbare [82] and Baifeng B, an indica landrace [83]. Similarly, miR528 which is highly expressed in Nipponbare grains [81], is also highly expressed in both SN and LGR (Additional file 2: table S14). Another case is of miR396, which is a negative regulator of GS in different genetic backgrounds in both indica and japonica rice [84]. Hence, it will be interesting to extrapolate the information generated in the present study to other genotypes and identify molecular markers of rice grain development and GS control.
Transcriptome changes regulating GS
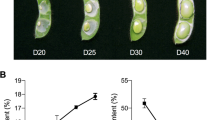
Transcriptome transitions control developmental progression. The morphological data of seeds and endosperm sections showed the full seed size and weight were obtained later in LGR (Fig. 1, Additional file 1: Figure S1). Developmental events also occurred later in LGR seed development. Hence, it can be hypothesized that slower progression of seed development might positively affect GS. To elucidate genes/processes responsible for this, the transcriptomes of SN and LGR during seed development were compared. Amongst the pathways with ≥ 50 genes with log2fc ≥ 10 (Additional file 1: Figure S4B), AGPs (under cell wall category) and GDSL motif lipases were more in LGR. AGPs are glycoproteins present on the cell wall and function in plant growth and development, including cell division and expansion. They express in various rice tissues, including seeds and seedling that are subjected to rapid changes in cell morphology [85, 86]. GDSL are multifunctional enzymes involved in fatty acid metabolism during seed germination and have been found to be downregulated in RGE1 mutants of Arabidopsis which exhibit smaller seeds [87, 88]. Abundance of these proteins in LGR might indicate their involvement in cell size increment. SSPs and LEAs were abundant in both SN and LGR. This is because SSPs are second major storage products in rice [14, 89]. LEA proteins form up to 4% of total cellular proteins in seeds and are associated with imparting drought tolerance during seed drying phase [90, 91]. The comparative transcriptome data shows an insight into the genes and pathways that might be responsible for GS increment.
LGR S3 stage is important for increase in GS
CS and SS pair-wise comparisons for oppositely regulated DEGs, categorized into the same pathways (Fig. 2D). This could imply that similar functions could be modulated by different genes in the two genotypes. LGR had higher genes in the DNA synthesis category. DEGs had an LGR-specific enrichment of cell cycle and DNA metabolism (Additional file 1: Figure S6A), and the number of cell cycle-related DEGs were highest for S3 stage of LGR (Fig. 3B). This was the stage where the endosperm cell size increased in LGR (Fig. 1I, xv, xvi), unlike SN. DEGs preferential to LGR S3 (Additional file 1: Figure S15) had a high number of cell cycle-related genes. Also, 38 LGR S3 stage-specific or preferential DEGs were located within QTLs governing GS. S3 stage in LGR overlapped with endoreduplication phase during cereal seed development [92, 93]. Expression of endoreduplication-related genes peaked in LGR S3 stage (Additional file 1: Figure S6C). Direct correlations exist between endoreduplication and cell size in plants [94]. It is known that endoreduplication increases cell size by increasing nuclear volume, driving cells to increase cell volume to maintain nucleo-cytoplasmic ratio [95], and by increasing gene expression to enhance reserve accumulation [96]. Hence, it can be hypothesized that higher endoreduplication levels in LGR might be positively contributing to GS, which can be verified further.
The process of grain filling, once completely understood, can be used to increase yield [97]. After starch, the second most abundant nutrient component of rice grain are SSPs [98]. Quality of SSPs and starch have a direct influence on each other [99, 104] and cellulose synthesis to mediate organ elongation in plants under different stimuli [105, 106]. Phosphorylation of fructose after cleavage of sucrose by fructokinases drives carbon metabolism towards starch synthesis and respiration. OsFKII, a rice fructokinase gene, expresses at high levels in endosperm [107] suggesting involvement in starch accumulation. Mutation in rice plastid phosphorylase gene, Pho1, reduces starch synthesis and causes abnormal seed morphology [108]. There are innumerable examples to show that GS and starch content have a direct correlation and starch has a forbearance on grain quality [109,110,114]. Hence, the genes related to carbohydrate biosynthesis and metabolism and SSP biosynthesis play an important role in control of GS and should be targeted for crop improvement through molecular breeding.
Regulators of GS
TFs regulate grain size and shape [55]. When the genes participating in/coding for TFs, hormone-related, cell cycle and growth, SSPs and carbohydrate pathway (Additional file 1: Figure S15) were sorted on the basis of their differential expression or specificity, amongst the seed-specific genes commonly expressed in all seed stages of SN and LGR, maximum genes were TFs indicating their regulatory importance during seed development. TF families (≥ 10 members) included Myb, AP2, NAC, zfC2H2, bZIP, Homeobox, MADS, B3, PHD, and Aux/IAA, in that order. For DEGs with log2fc ≥ 10, TF encoding genes were most abundant, again, highlighting their regulatory importance. Genes from many of these families regulate seed development [12, 37, 38, 51, 53]. Comparison of expression of TFs between SN and LGR showed that members of bZIP, NF-YC, C2H2, GATA, MADS, FHA, NF-YB, PHD, LBD, PLATZ, SBP, WRKY, and DOF families might regulate GS in various capacities and are also located within QTLs (Fig. 6, Additional file 2: Table S11). Of these, RISBZ1, a bZIP TF is involved in starch synthesis in rice seeds [115]. bZIPs regulate amylose biosynthesis in wheat grain [116]. NF-Ys regulate rice grain quality [117]and control endosperm development [118]. OsNF-YB1 regulates nutrient transport via sucrose transporters in endosperm during grain filling [81]. Conserved miRNAs included osa-miR529 and osa-miR396, which are involved in seed size regulation [151, 152]. In the contrasting set of miRNAs downregulated in all stages of LGR, several miRNAs associated with seed size and grain filling were present, including osa-miR166 [153], osa-miR398, and osa-miR159 [154]. Few of the targets were cyclin-related proteins (targeted by osa-miR167i-3p) and FtsZ2-1 (targeted by osa-miR172a). OsFtsZ2 is homologous to bacterial cytokinesis-related FtsZ and is implicated positively in amyloplast division in rice [44]. There was also an expansin precursor, which mediates cell wall expansion [155]. These miRNAs are downregulated throughout seed development in LGR to enhance GS by promoting numerous aspects of seed development, including cell proliferation, cell elongation, energy production, and grain filling [9, 44, 96, 134]. Thus, new potential miRNA-target modules identified here should be explored further to establish their roles in GS regulation.
miRNA-target pairs with similar differential regulation in both SN and LGR
miRNA-target modules with similar regulation throughout both SN and LGR (Fig. 8) can be pivotal regulators of seed development. miR396a-3p is one such molecule. There are 9 members in osa-miR396 family. Of these, miR396c, miR396e, and miR396f are negative regulators of grain length, width, and weight [158]. MIM396-5p plants, where all of the above are downregulated, have increased grain length but decreased grain width [84]. Grains of Baifeng B (an indica landrace) and japonica cultivar Zhongua 11 show high expression of miR396 [57, 83]. In SN and LGR, miR396a-3p was upregulated throughout grain development, though to a higher extent in SN (Fig. 8A). Its putative target is novel. Hence, miR396a-3p might be an important regulator of rice seed development and should be explored further. Also, in Arabidopsis leaf, the levels of miR396 limit cell proliferation [159], indicating a similar role in grain. Additionally, in our data, 13 miRNAs belonging to this family (including -3p and -5p forms) are differentially expressed, and regulation of GS might be a combination of these. miR396 functions through miR408, a positive regulator of grain size [84]. In concordance, both miR408-5p and miR408-3p were downregulated throughout grain development in SN and LGR (Fig. 8B). However, it is known that osa-miR408-3p targets constitutively expressed BRD2/LTBSG1 and is involved in BR-mediated cell elongation in rice seeds [160]. Often genes regulate multiple aspects of rice grain development, including grain size, starch, and seed storage protein biosynthesis. Since miR396/miR408 module is differentially regulated throughout seed development, and with novel targets, it is possible, these miRNA-target pairs are major regulators of rice grain development, and need to be explored further. In addition, osa-miR319b which was commonly upregulated during seed development in SN and LGR (Fig. 8A) is known to target OsCAF2, which regulates normal chloroplast development [161]. This suggests suppression of chloroplast development in rice seeds. miR319 has been shown to target TCPs in leaf development [162]. It is highly upregulated in our data (Fig. 8A), and with novel targets, hinting at new avenues to explore. miR444 is known to regulate tillering and ovule development [163, 164]. It is downregulated in both SN and LGR (Fig. 8B) though to varying extents. miR528 is known to control flowering time, pollen formation, and plant height [171, 172]. GPDH is involved in energy production from gluconeogenesis. Inhibition of sucrose production via gluconeogenesis triggers compensated cell enlargement in Arabidopsis cotyledons [173], and hence, these targets might have a role in GS regulation. Four members of miR2118 family and their targets were oppositely regulated between SN and LGR in either CS or SS stages (Fig. 9). miR2118 is essential for proper reproductive development by formation of anther wall [174]. Since its members are upregulated in LGR stages, this family can be studied further for their role in GS control.
Briefly, it appears that genes associated with BR signaling, cell expansion, and stress tolerance experience opposite regulation, via miRNAs, in SN and LGR. Furthermore, miRNAs with opposite regulation between SN and LGR, seem to favor cell enlargement by modulating BR signaling and lipid metabolism [168, 173], thereby increasing GS in LGR. In addition, the localization of these targets on QTLs associated with GS strengthens the probability of their involvement in the process. Thus, novel miRNA-target modules identified for SN and LGR can provide suitable avenues for future studies in rice GS regulation.
The Domino effect model of seed size regulation
This extensive comparative morphological, histological, and transcriptome analysis of SN and LGR seeds throughout seed development concludes with a “Domino effect” model of seed size regulation (Fig. 10), emphasizing significance of the chronology of seed developmental events in governing GS. Just as falling of one domino triggers the next one, completion of one seed development event initiates the next. This process is strictly overseen by TFs, hormonal interplay, and miRNA regulation. Stage-wise comparison of the transcriptome of five seed stages showed that any given stage of LGR was most similar to the preceding stage in SN, also validated by endosperm sections. Delayed cellularization in LGR indicated a longer period of initial cell division phase (Figs. 1 and 2), supported not only by enrichment of cell cycle-related genes in its transcriptome, but also continuation of cell cycle till S3 stage (Fig. 3). Maximum increment in seed weight in SN and LGR occurred in S3 and S4 stages, respectively, indicative of delayed storage reserve accumulation in LGR (Figs. 1H, 4 and 5), as a consequence of predominant expression of carbohydrate and SSP biosynthetic genes from SN S2 and LGR S3 stages onwards. Storage reserve accumulation occurs after cellularization [13]and enhances seed size and weight [23, 119]. As cellularization is prolonged in LGR, accumulation of storage compounds is procrastinated (Fig. 10). Cells in LGR spikelets were larger in size (Fig. 1; Additional file 1: Figure S1A), suggesting enhanced cell expansion. Subsequently, enhanced endosperm cell size and prominent nuclei were apparent in S3 stage, marking it as the period of cell elongation, coinciding with endoreduplication phase [12]. Markers of endoreduplication showed peak expression at LGR S3 (Additional file 1: Figure S6C). Additionally, LGR S3 transcriptome was most unique in comparison with other stages (Figs. 2B and 3), signifying intense transcriptome reprogramming to increase seed size. Thus, larger seed size in LGR appears to be a result of enhanced cell expansion via endoreduplication during S3 stage (Fig. 10). Lastly, PCD started early in SN endosperm cells (Fig. 1I and Additional file 1: Figure S1C), induced probably by expression of positive regulators of PCD from S2 stage onwards (Additional file 1: Figure S8B and C). Precocious cellularization and PCD are known to reduce GS in rice [25, 26], thus restricting GS in SN, and in turn supporting the postulated “Domino effect”. Essential events occurring during seed development, namely cell cycle, cell expansion, storage accumulation, and PCD, appear to be modulated by temporal regulation of phytohormones in the two genotypes creating differences in GS (Figs. 7 and 10). Moreover, extensive miRNA regulation of genes throughout seed development (Fig. 8) adds another level of regulation in this process. Our study also suggests the presence of new miRNA-target modules that need to be functionally validated for their roles in rice seed development. The postulated Domino effect model is also supported by the transcriptome analyses of seed development in IR64, which has a medium-sized grain [14]. Thus, a “Domino effect” influences seed development wherein one process/pathway is overlapped by the next one, and it is the extent of one process that determines the occurrence of subsequent one, thereby regulating seed size.

The “Domino effect” model of seed size regulation. Diagram represents progression of the major seed developmental events in SN and LGR. Upper and lower panels represent SN and LGR, respectively (as mentioned on the left of the diagram). S1–S5 represent five stages of seed development. Orange, red, green, violet, and blue bars represent progression of cell cycle, cellularization, endoreduplication, storage reserve accumulation, and PCD during seed development (as indicated in the color legend). Lines represent accumulation pattern of hormones during seed development in SN and LGR based on genes showing different expression pattern in the two genotypes. Pathways specific or preferential to S3 stage in LGR contributing to enhanced cell size have been mentioned between the dotted red lines below S3 stage. Comparable seed developmental stages have been connected with dotted gray lines
Conclusions
Comparison of transcriptome of five seed development stages from SN and LGR highlights the importance of S3 (5–10 DAP) stage in LGR, for increment of rice grain size. S3 stage of LGR has the most unique transcriptome amongst all comparisons. This is the stage where maximum number of cell cycle genes specifically express, and the increment in total protein content is highest. All events of seed development, including grain filling, occur later in LGR. Genes involved in phytohormone pathways (136 genes) and members from nine transcription factor families contributing to temporal changes have been elucidated. The DEGs underlying the QTLs will have functional relevance for genetic dissection of GS trait in rice. Novel miRNA-target pairs which might contribute to seed development or GS increment have been determined. Out of these five miRNAs show upregulation throughout seed development in both SN and LGR and target nine genes. Also, five miRNAs are downregulated throughout seed development in both SN and LGR and target eight genes. Eight miRNAs and their nine targets have opposite regulation between SN and LGR and could potentially regulate GS. The analyses have led us to propose a “Domino effect” model for rice grain increment. In this, the attainment of completion of one step of grain development triggers the next one. Since each event is slower in LGR, there is a temporal lag leading to increased cell size and subsequently higher grain filling, and eventually bigger grain size. The expression data for all genes and miRNAs from this study are available to RGDD. Availability of such information on a single platform will not only be useful for rice yield enhancement but can be extrapolated to other crops as well.
Methods
Plant materials
Long-grained and short-grained indica rice, LGR [66], and Sonasal (SN), respectively, were grown in the kharif season in the field conditions at NIPGR, New Delhi, India. Once panicle emergence started, individual panicles were observed for anthesis. Panicles took 3–4 days to complete emergence and anthesis. Each panicle on the plant was tagged on the day of its anthesis (by mentioning the date) before noon, particularly the region where freshly dehisced anthers are visible, as the process follows a basipetal direction. The pollinating spikelets on the lowermost part of the panicle were often left untagged, due to staggered anthesis. The day of anthesis was counted as 0 DAP, as pollination occurs in a few hours. The tagged panicles were left on the plant to mature. Individual seeds were separated and collected for each DAP, as they matured and harvested in liquid nitrogen. While harvesting, empty seeds were discarded. Seeds from each DAP were stored at − 80 °C. At the time of RNA isolation, equal weights of seeds for each DAP were pooled according to the categories, S1 (0–2 DAP), S2 (3–4 DAP), S3 (5–10 DAP), S4 (11–20 DAP), and S5 (21–29 DAP). Each biological replicate was made from a separate pool of seeds. Dried mature seeds of SN and LGR were harvested for estimation of grain length, width, and weight.
Seed trait measurements
Freshly harvested seeds of 0, 1, 2, 3, 4, 7, 10, 13, 18, 21, and 29 DAP were collected in 30% ethanol. Images of husked and dehusked seeds of SN and LGR were captured at × 4 magnification under a stereozoom microscope (AZ100, Nikon; Japan). Grain length and width were quantified per seed by taking average of 100 seeds using WinSEEDLE™ software (Regent Instruments Inc.; Canada). One thousand-grain weight was measured in biological triplicates. Grain filling rate was estimated by measuring fresh weight of 15 seeds in triplicates from each DAP after removing stalks and awns. The measurements from each DAP constituting a stage were added to obtain the weight of 15 seeds/stage.
SEM analysis of spikelet
Freshly harvested spikelets belonging to 7 DAP harvested from SN and LGR were immersed completely in FAE fixative solution [10% formaldehyde, 5% acetic acid, and 50% absolute ethanol] in 15 ml Borosil® glass vials. These samples were vacuum infiltrated for 30 min and incubated at 4 °C overnight in dark. The tissue samples were subjected to dehydration series with increasing gradient of ethanol (60%, 70%, 80%, 95%, and 100%) at room temperature for 1 h. The outer surface cells of lemma were observed under EVO® LS10 scanning electron microscope (ZEISS, Germany) at × 500 magnifications. The images were analyzed using ImageJ [175] to estimate cell length and cell area of the outer epidermal cells in the middle region of the lemma.
Histological study
Freshly harvested seeds of 4, 5, 7, 9, and 11 DAP were dehusked, fixed, and dried in ethanol series (as mentioned in the previous section). Ethanol was gradually replaced with xylene by kee** the tissues in increasing concentration of xylene and decreasing concentration of ethanol in the percentages of 25:75, 50:50, and 75:25. Next, two rounds of treatment with 100% xylene were given to the seeds at room temperature for 1 h on the rocker, followed by infiltration with paraplast X-TRA (Sigma-Aldrich; USA) by adding molten paraplast every 2–3 h at 60 °C for 3 days. The wax-infiltrated tissues were then embedded into molds (Yorko®; India), and 10-μm sections were cut for 4, 5, and 7 DAP and 15-μm sections were cut for 9 and 11 DAP using rotary microtome (Leica Biosystems, Germany). Paraplast was removed from the sections by immersing the slides in HistoChoice® clearing agent (Sigma-Aldrich; USA) for 1 h. Sections were stained with 0.01% toluidine blue-O for 10 min or 0.1% Coomassie brilliant blue/CBB [0.25% CBB in 50% methanol and 10% acetic acid] for 20 min or 2% I2/KI solution (2 g KI and 0.2 g Iodine in 100 ml MQ) for 2–5 min. The sections were mounted using D.P.X. mountant (Himedia®, India) and were visualized under light microscope (Eclipse 80i, Nikon; Japan) at × 20 magnification.
Total RNA isolation from seed tissue and flag leaf
Total RNA was isolated from five seed developmental stages of the two rice genotypes LGR and SN, namely, S1 (0–2 DAP), S2 (3–4 DAP), S3 (5–10 DAP), S4 (11–20 DAP), and S5 (21–29 DAP) using a seed-specific protocol [176] with few modifications, as previously described [37, 52, 80, 177]. Briefly, 100 mg tissue was taken by pooling equal amounts of seed tissue from the respective DAP constituting a stage. The tissue was ground to a fine powder in liquid nitrogen and mixed with extraction buffer [50 mM Tris–HCl pH 9.0, 20 mM EDTA pH 8.0, 150 mM NaCl, 1% N-lauryl sarcoyl (sodium salt), and 5 mM DTT], followed by phenol:chloroform:isoamyl alcohol (25:24:1) treatment. Then, GH buffer [8 M guanidine hydrochloride, 20 mM 2-[N-Morpholino] ethanesulfonic acid, 0.5 M EDTA, 50 mM β-mercaptoethanol] and phenol:chloroform:isoamyl alcohol were added to the supernatant, followed by chloroform treatment. RNA was precipitated by adding 3 M sodium acetate (pH 5.2) and twice volume of chilled ethanol. The pellets were washed twice with 70% ethanol, dried, and dissolved in 40 μl DEPC-treated deionized water. RNA from flag leaves of SN and LGR were isolated using TRI Reagent® Solution (Invitrogen™; USA) as per manufacturer’s protocol. DNase treatment was given to the RNA samples using RNase-Free DNase Set (Qiagen; Germany). The purity and concentration of RNA samples were checked by NANODROP 2000c Spectrophotometer (Thermo Scientific; USA) and on 1% denaturing agarose gel in 1 × MOPS buffer [400 mM MOPS, 99.6 mM sodium acetate, 20 mM EDTA] with 1.1% formaldehyde. Integrity of RNA samples was checked using 1 μl RNA sample (25–500 ng/μl) by Agilent 2100 Bioanalyzer.
RNA-Seq library preparation and sequencing
The RNA isolated from the seed and the leaf samples (in biological triplicates) were used for cDNA library preparation according to the TrueSeq® RNA Sample Preparation v2 Guide (Illumina®; USA) according to the manufacturer’s protocol as described previously [53, 80]. Briefly, poly-A mRNA purified using oligo(dT)-attached magnetic beads were fragmented and primed, followed by double-stranded cDNA synthesis using SuperScript II Reverse transcriptase (Invitrogen™; USA), Second Strand Master Mix, End Repair Mix and Resuspension Buffer. Blunt-ended ds cDNA fragments were then adenylated and adapters were ligated. Following this, DNA fragments were enriched by PCR and paired-end sequencing was performed using Illumina HiSeq™ 2000.
Whole transcriptome sequencing data analysis
Data analysis was done as mentioned earlier [53, 80]. Low-quality reads were trimmed using Cutadapt (v1.8.1) (Martin, 2011). The unwanted sequences, including non-polyA tailed RNA, rRNAs, tRNAs, adapter sequences, mitochondrial genome, were removed using Bowtie2 (v2.1.0) (Langmead, 2010), in-house perl scripts and picard tools (v1.85). The clean reads thus obtained were used for expression and differential expression analysis. The clean reads were aligned to the reference genome (MSU 7) by TopHat (v2.0.8) [178]. The uniquely mapped reads were used for estimation of gene expression using Cufflinks program (v2.0.2) [179]. Pearson’s correlation coefficient and PCA between the biological replicates was calculated and visualized using corrplot and pca3d packages of R (version 3.2.0; https://cran.r-project.org/) to estimate the relatedness between the biological replicates and the tissue samples. The normalized gene expression data was represented as FPKM (fragments per kilobase per million of reads mapped). All genes with an FPKM ≥ 1 were counted as expressed and considered for downstream analyses after removal of transposable element (TE)-related genes. Differential expression in the seed developmental stages was calculated by Cuffdiff program (v2.0.2) [180] with respect to flag leaf at p-value ≤ 0.05 and q-value ≤ 0.05. DEGs during seed development in SN and LGR were determined with respect to flag leaf (as vegetative control) from each genotype. Stringent cutoffs of FPKM value ≥ 1, log2fold change (log2fc) value ≥ 1 and q-value ≤ 0.05 were used for identification of DEGs. GO enrichment analysis of the expressed and DEGs was performed using agriGO software (v1.2) [181] and BiNGO plugin [182] of Cytoscape (version 3.4.0). Heatmaps and k-means cluster diagrams were prepared using MeV_4_6_0 [183] employing Euclidean distance method. Pathway annotations were performed using MapMan software (version 3.6.0RC1) [184]. Functional annotation of genes was performed using RGAP version 7 and funRiceGenes (https://funricegenes.github.io/). Bubble plots were prepared in R software using ggplot2 (Wickham, 2016) and reshape2 [185] packages.
Real-time PCR validation
cDNA for qPCR validation were prepared from the RNA isolated from the five stages of seed development and flag leaf in two biological replicates as detailed previously, using Superscript™ III first-strand cDNA synthesis kit (Invitrogen™; USA) [37, 52, 53, 80, 177]. Primers for real-time PCR were made from the unique regions of the selected genes using Primer Express 3.0. miRNA sequence was taken from miRBase (http://mirbase.org). The assay was carried out with Fast SYBR® Green Master Mix (Applied Biosystems; USA) as mentioned previously [177]. Real-time PCR was done on 7500 Fast Real-Time System (Applied Biosytsems; USA), and 7500 software v2.0.1 was used for data analysis. Housekee** gene, OsACT1, was used as the endogenous control for real-time PCR. Fold change was calculated by ΔΔCT method. List of primers has been given in Additional file 2: Table 17.
QTL map**
Molecular map** of QTLs was performed to establish the correspondence amongst grain size/weight QTLs with the genes having pronounced expression especially during seed developmental stages in rice. For this, SNPs exhibiting differentiation between high (40 g) and low (10 g) 1000-grain weight parental genotypes of a F5 map** population (LGR × Sonasal) were genotyped using the genomic DNA of 286 map** individuals using the Illumina Infinium assay (https://www.illumina.com). The SNP genoty** information showing goodness-of-fit towards the expected Mendelian 1:1 segregation ratio was analyzed using the JoinMap 4.1 (http://www.kyazma.nl/index.php/mc.JoinMap) at a higher LOD threshold (3.0) with Kosambi map** function. SNPs were mapped into defined linkage groups/chromosomes according to their centiMorgan (cM) genetic distances and physical positions (bp). The individuals along with parents of a map** population were phenotyped for 2 years, for grain size/weight grown in the field at NIPGR, New Delhi. To identify grain size/weight QTLs, the genoty** data of SNPs mapped on 12 rice chromosomes was correlated with grain size/weight trait phenotypic data of map** individuals and parental genotypes using composite interval map** function of MapQTL 6 at a LOD threshold score > 2.5 with 1000 permutations (p < 0.05 significance). The phenotypic variation explained by each major grain size/weight QTL at a significant LOD was estimated. Further, the genes showing high differential expression in SN and LGR in each analysis were delineated. The physical positions of these genes were matched with the QTL genomic regions. The ones overlap** with a QTL were separated.
Small RNA library preparation
Total RNA was isolated from the five seed developmental stages and flag leaf tissue of SN and LGR. cDNA library was prepared according to the TrueSeq™ Small RNA library preparation kit (Illumina®; USA). Briefly, adapters were ligated sequentially at 3′ and 5′ end of the RNA, respectively. cDNA was prepared to selectively enrich the adapter ligated RNA fragments by performing PCR with two primers that anneal to the ends of the adapters. Next, the cDNA prepared was amplified by PCR and indexed with RNA PCR primer 1 (RP1) and RNA PCR primer Index (RPIX), respectively. The cDNA libraries were gel purified, and the quality of the libraries was checked by running 1 μl of the products on Agilent tape station with DNA HS Screen tape. The obtained libraries were then normalized to 2 nM concentration for cluster generation on Illumina sequencing platforms by adding Tris HCL 10 mM, pH 8.5. Following this, single-end sequencing was performed using Illumina MiSeq® system.
Small RNA sequencing and data analysis
Low-quality reads were trimmed using Cutadapt (v1.3) (Martin, 2011). The adapter removed reads were aligned against siRNA, snRNA, snoRNA, tRNA, and rRNA using nc-RNA databases (siRNAdb; http://sirna.sbc.su.se/sirnadb_050915.txt; NCBI Genbank; http://www.ncbi.nlm.nih.gov/genbank/; deepBase; http://deepbase.sysu.edu.cn/download.php; GtRNAdb; http://gtrnadb.ucsc.edu/; Rfam; http://rfam.xfam.org/) with Bowtie2 program v2.1.0 [186]. The unaligned clean reads of 17–35 bp length were used for miRNA identification by aligning to mature miRNAs of Oryza sativa in miRBase release-21 (http://www.mirbase.org/) using Bowtie program (version 0.12.9). The expression data of miRNAs were normalized using the TPM (transcripts per million) method. The differential expression analysis of the miRNAs was estimated using a negative binomial method with the DESeq package [187] of R (https://cran.r-project.org/) with default parameters. The targets of the known miRNAs were identified using psRNA Target [188] tool with default parameters.
Stem-loop qRT-PCR
Stem-loop qRT-PCR was done to detect the levels of miR530-5p in SN and LGR seed developmental stages (S1–S5). Total RNA was isolated from S1–S5 stages and flag leaf of SN and LGR as mentioned above. The total RNA was purified using acid phenol (Ambion®; Naugatuck, CT, USA) as per manufacturer’s protocol. For stem-loop qRT-PCR, miR530-5p specific cDNA was synthesized using its specific stem-loop reverse transcription primer (miR530-5p SL primer) (Additional file 2: table 17). This cDNA was synthesized from 200 ng of total RNA by using Superscript™ III first-strand cDNA synthesis kit (Invitrogen™; USA) according to the manufacturer’s protocol. This cDNA was used for qRT-PCR assay using miR530-5p forward primer and a universal reverse primer. The conditions for qRT-PCR were maintained same as mentioned previously. snRNA U6 was used as an internal control. Three biological replicates were used for the assay and significance was calculated using Student’s t test with p value˂0.001 denoted as double asterisk (**).
Total starch and protein isolation and quantification
A total of 100 mg of develo** rice seeds from each of S1–S5 stages of SN and LGR were finely ground to powder in liquid nitrogen and incubated for 10 min at 4 °C after homogenizing with 1 mL of lysis buffer (20 mM Tris pH 7.6, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF). The insoluble debris was removed from the mixture by spinning at 100 g for 10 min at 4 °C. Supernatant was centrifuged at 12,000 g for 10 min at 4 °C to obtain the soluble protein fraction [189]. Total isolated proteins from each sample were quantified according to the Bradford method [190] (Amresco M173-KIT).
Seeds from all developmental stages (S1–S5) of SN and LGR were crushed using liquid nitrogen. One hundred milligrams of the crushed sample was used for isolating starch. Starch isolation and determination was done by using starch assay kit (Megazyme, Wicklow, Ireland, http://www.megazyme.com/), according to the manufacturer’s protocol. Starch estimation was done in two independent biological replicates.
Database development
RGDD has been developed using Tableau Software (https://www.tableau.com/), which is a leading data visualization software used for reporting and analyzing vast volumes of data. The background data input for this database was in CSV format. It was obtained subsequent to the abovementioned transcriptome and miRNome analyses. The programming was done within Tableau Software itself.
Availability of data and materials
All data generated or analyzed during this study are included in this published article, its additional files and publicly available repositories.
High-throughput RNAseq data has been deposited in the Gene Expression Omnibus (GEO) database under the SRA accession number PRJNA605919, ID 605,919—BioProject—NCBI (nih.gov). The small RNAseq data has been deposited under SRA accession number PRJNA616068, ID 616,068—BioProject—NCBI (nih.gov). The analyzed data can be accessed at the Rice Grain Development Database at www.nipgr.ac.in/RGDD/index.php.
Abbreviations
- ABA:
-
Abscisic acid
- AGPs:
-
Arabinogalactan proteins
- BR:
-
Brassinosteroid
- CBB:
-
Coomassie Brilliant Blue
- CK:
-
Cytokinin
- cM:
-
Centi Morgan
- CS:
-
Comparable stages
- DAP:
-
Days after pollination
- DEGs:
-
Differentially expressed genes
- DEMs:
-
Differentially expressed miRNAs
- ETC:
-
Electron transport chain
- FPKM:
-
Fragments per kilobase per millions of reads mapped
- GA:
-
Gibberillic acid
- GDSL:
-
Gly-Asp-Ser-Leu
- GO:
-
Gene ontology
- GS:
-
Grain size
- JA:
-
Jasmonic acid
- LEA:
-
Late embryogenesis abundant proteins
- LGR:
-
Large grained rice
- LOD:
-
Logarithm of the odds
- PCA:
-
Principal component analysis
- PCD:
-
Programmed cell death
- PHD:
-
Plant homeo domain
- QTL:
-
Quantitative trait locus
- RGAP:
-
Rice Genome Annotation Project
- RGDD:
-
Rice Grain Development Database
- RP1:
-
RNA PCR Primer1
- RPIX:
-
RNA PCR Primer Index
- SA:
-
Salicylic acid
- SEM:
-
Scanning electron microscopy
- SN:
-
Sonasal
- SNPs:
-
Single-nucleotide polymorphisms
- SS:
-
Same stages
- SSP:
-
Seed storage protein
- TAG:
-
Triacyl glycerol
- TCA:
-
Tricarboxylic acid
- TE:
-
Transposable element
- TFs:
-
Transcription factors
- TPM:
-
Transcripts per million
- TT:
-
Tai tobacco
- UGT:
-
UDP glucosyl and glucoronyl transferase
- YT:
-
Yan tobacco
References
Li Y, **ao J, Chen L, Huang X, Cheng Z, Han B, et al. Rice functional genomics research: past decade and future. Mol Plant. 2018;11(3):359–80.
Bailey-Serres J, Parker JE, Ainsworth EA, Oldroyd GED, Schroeder JI. Genetic strategies for improving crop yields. Nature. 2019;575(7781):109–18.
Coulter JK. World agriculture: towards 2015/2030. An FAO perspective. Edited by J. Bruinsma. Rome: FAO and London: Earthscan; 2003. pp. 432. ISBN 92-5-104835-5.
Huang R, Jiang L, Zheng J, Wang T, Wang H, Huang Y, et al. Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci. 2013;18(4):218–26.
Fitzgerald MA, McCouch SR, Hall RD. Not just a grain of rice: the quest for quality. Trends Plant Sci. 2009;14(3):133–9.
Wang A, Hou Q, Si L, Huang X, Luo J, Lu D, et al. The PLATZ transcription factor GL6 affects grain length and number in rice. Plant Physiol. 2019;180(4):2077–90.
Yuan H, Gao P, Hu X, Yuan M, Xu Z, ** M, et al. Fine map** and candidate gene analysis of qGSN5, a novel quantitative trait locus coordinating grain size and grain number in rice. Theor Appl Genet. 2022;135(1):51–64.
Si L, Chen J, Huang X, Gong H, Luo J, Hou Q, et al. OsSPL13 controls grain size in cultivated rice. Nat Genet. 2016;48(4):447–56.
Liu X, Wei R, Sheng Z, Jiao G, Tang S, Luo J, et al. Polycomb protein OsFIE2 affects plant height and grain yield in rice. PloS one. 2016;11(10):e0164748.
Coventry S, Barr AR, Eglinton JK, McDonald GK. The determinants and genome locations influencing grain weight and size in barley (Hordeum vulgare L.). Crop Pasture Sci. 2003;54:1103–15.
Zhao D-S, Li Q-F, Zhang C-Q, Zhang C, Yang Q-Q, Pan L-X, et al. GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nat Commun. 2018;9(1):1240.
Agarwal P, Kapoor S, Tyagi AK. Transcription factors regulating the progression of monocot and dicot seed development. BioEssays. 2011;33(3):189–202.
Wu X, Liu J, Li D, Liu C-M. Rice caryopsis development II: Dynamic changes in the endosperm. J Integr Plant Biol. 2016;58(9):786–98.
Sharma R, Agarwal P, Ray S, Deveshwar P, Sharma P, Sharma N, et al. Expression dynamics of metabolic and regulatory components across stages of panicle and seed development in indica rice. Funct Integr Genomics. 2012;12(2):229–48.
Zhou Y, Tao Y, Zhu J, Miao J, Liu J, Liu Y, et al. GNS4, a novel allele of DWARF11, regulates grain number and grain size in a high-yield rice variety. Rice. 2017;10(1):34.
Sun S, Wang L, Mao H, Shao L, Li X, **ao J, et al. A G-protein pathway determines grain size in rice. Nat Commun. 2018;9(1):851.
Yang W, Wu K, Wang B, Liu H, Guo S, Guo X, et al. The RING E3 ligase CLG1 targets GS3 for degradation via the endosome pathway to determine grain size in rice. Mol Plant. 2021;14(10):1699–713.
Biswal AK, Wu TY, Urano D, Pelissier R, Morel JB, Jones AM, et al. Novel mutant alleles reveal a role of the extra-large G protein in rice grain filling, panicle architecture, plant growth, and disease resistance. Front Plant Sci. 2021;12:782960.
Li J, Chu H, Zhang Y, Mou T, Wu C, Zhang Q, et al. The rice HGW gene encodes a ubiquitin-associated (UBA) domain protein that regulates heading date and grain weight. PLoS ONE. 2012;7(3):e34231.
Gao Q, Zhang N, Wang WQ, Shen SY, Bai C, Song XJ. The ubiquitin-interacting motif-type ubiquitin receptor HDR3 interacts with and stabilizes the histone acetyltransferase GW6a to control the grain size in rice. Plant Cell. 2021;33(10):3331–47.
Wang E, Wang J, Zhu X, Hao W, Wang L, Li Q, et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet. 2008;40(11):1370–4.
Sun T, Li S, Ren H. OsFH15, a class I formin, interacts with microfilaments and microtubules to regulate grain size via affecting cell expansion in rice. Sci Rep. 2017;7(1):6538.
Zhang H, Lu Y, Zhao Y, Zhou DX. OsSRT1 is involved in rice seed development through regulation of starch metabolism gene expression. Plant Sci. 2016;248:28–36.
Eom J-S, Cho J-I, Reinders A, Lee S-W, Yoo Y, Tuan PQ, et al. Impaired function of the tonoplast-localized sucrose transporter in rice, OsSUT2, limits the transport of vacuolar reserve sucrose and affects plant growth. Plant Physiol. 2011;157(1):109–19.
Chen C, Begcy K, Liu K, Folsom JJ, Wang Z, Zhang C, et al. Heat stress yields a unique MADS box transcription factor in determining seed size and thermal sensitivity. Plant Physiol. 2016;171(1):606–22.
Zhang L, Jiang D, Pang J, Chen R, Wang X, Yang D. The endoplasmic reticulum stress induced by highly expressed OsrAAT reduces seed size via pre-mature programmed cell death. Plant Mol Biol. 2013;83(1–2):153–61.
Fu JR, Zhu LX, Sun XT, Zhou DH, Ouyang LJ, Bian JM, et al. Genetic analysis of grain shape and weight after cutting rice husk. Genet Mol Res. 2015;14(4):17739–48.
Sabelli PA, Larkins BA. The development of endosperm in grasses. Plant Physiol. 2009;149(1):14–26.
Itoh J, Nonomura K, Ikeda K, Yamaki S, Inukai Y, Yamagishi H, et al. Rice plant development: from zygote to spikelet. Plant Cell Physiol. 2005;46(1):23–47.
Jiao Y, Tausta SL, Gandotra N, Sun N, Liu T, Clay NK, et al. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat Genet. 2009;41(2):258–63.
Fujita M, Horiuchi Y, Ueda Y, Mizuta Y, Kubo T, Yano K, et al. Rice expression atlas in reproductive development. Plant Cell Physiol. 2010;51:2060–81.
Mahto A, Mathew I, Agarwal P. Decoding the transcriptome of rice seed during development. In: Jimenez-Lopez JC, editor. Advances in Seed Biology. Spain: InTech; 2017. p. 25.
Huh SM, Hwang YS, Shin YS, Nam MH, Kim DY, Yoon IS. Comparative transcriptome profiling of develo** caryopses from two rice cultivars with differential dormancy. J Plant Physiol. 2013;170(12):1090–100.
Zhang F, Huang LY, Zhang F, Ali J, Cruz CV, Zhuo DL, et al. Comparative transcriptome profiling of a rice line carrying Xa39 and its parents triggered by Xanthomonas oryzae pv. oryzae provides novel insights into the broad-spectrum hypersensitive response. BMC Genom. 2015;16(1):111.
Wang J, Zhu J, Zhang Y, Fan F, Li W, Wang F, et al. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of indica rice at seedling stage. Sci Rep. 2018;8(1):2085.
Wei G, Tao Y, Liu G, Chen C, Luo R, **a H, et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc Natl Acad Sci USA. 2009;106(19):7695–701.
Agarwal P, Arora R, Ray S, Singh AK, Singh VP, Takatsuji H, et al. Genome-wide identification of C2H2 zinc-finger gene family in rice and their phylogeny and expression analysis. Plant Mol Biol. 2007;65(4):467–85.
Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007;8(1):242.
Sato Y, Antonio B, Namiki N, Motoyama R, Sugimoto K, Takehisa H, et al. Field transcriptome revealed critical developmental and physiological transitions involved in the expression of growth potential in japonica rice. BMC Plant Biol. 2011;11:10.
Nie DM, Ouyang YD, Wang X, Zhou W, Hu CG, Yao J. Genome-wide analysis of endosperm-specific genes in rice. Gene. 2013;530(2):236–47.
Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12(12):1871–83.
Dante RA, Larkins BA, Sabelli PA. Cell cycle control and seed development. Front Plant Sci. 2014;5:493.
Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7(3):254–61.
Xu SB, Li T, Deng ZY, Chong K, Xue Y, Wang T. Dynamic proteomic analysis reveals a switch between central carbon metabolism and alcoholic fermentation in rice filling grains. Plant Physiol. 2008;148(2):908–25.
Yang X, Wu F, Lin X, Du X, Chong K, Gramzow L, et al. Live and let die - the B(sister) MADS-box gene OsMADS29 controls the degeneration of cells in maternal tissues during seed development of rice (Oryza sativa). PLoS ONE. 2012;7(12):e51435.
Fan J, Bai P, Ning Y, Wang J, Shi X, **ong Y, et al. The monocot-specific receptor-like kinase SDS2 controls cell death and immunity in rice. Cell Host Microbe. 2018;23(4):498.
Dou M, Zhang Y, Yang S, Feng X. Identification of ZHOUPI orthologs in rice involved in endosperm development and cuticle formation. Front Plant Sci. 2018;9:223.
Huang L, Sun Q, Qin F, Li C, Zhao Y, Zhou D-X. Down-regulation of a SILENT INFORMATION REGULATOR2-related histone deacetylase gene, OsSRT1, induces DNA fragmentation and cell death in rice. Plant Physiol. 2007;144(3):1508–19.
Luo J, Liu H, Zhou T, Gu B, Huang X, Shangguan Y, et al. An-1 encodes a basic helix-loop-helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell. 2013;25(9):3360–76.
Nayar S, Sharma R, Tyagi AK, Kapoor S. Functional delineation of rice MADS29 reveals its role in embryo and endosperm development by affecting hormone homeostasis. J Exp Bot. 2013;64(14):4239–53.
Mathew IE, Agarwal P. May the fittest protein evolve: favoring the plant-specific origin and expansion of NAC transcription factors. BioEssays. 2018;40(8):e1800018.
Das S, Parida SK, Agarwal P, Tyagi AK. Transcription factor OsNF-YB9 regulates reproductive growth and development in rice. Planta. 2019;250(6):1849–65.
Mathew IE, Priyadarshini R, Mahto A, Jaiswal P, Parida SK, Agarwal P. SUPER STARCHY1/ONAC025 participates in rice grain filling. Plant Direct. 2020;4(9):e00249.
Seo M, Hanada A, Kuwahara A, Endo A, Okamoto M, Yamauchi Y, et al. Regulation of hormone metabolism in Arabidopsis seeds: phytochrome regulation of abscisic acid metabolism and abscisic acid regulation of gibberellin metabolism. Plant J. 2006;48(3):354–66.
Azizi P, Osman M, Hanafi MM, Sahebi M, Rafii MY, Taheri S, et al. Molecular insights into the regulation of rice kernel elongation. Crit Rev Biotechnol. 2019;39(7):904–23.
Li N, Xu R, Li Y. Molecular networks of seed size control in plants. Annu Rev Plant Biol. 2019;70:435–63.
Xue LJ, Zhang JJ, Xue HW. Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res. 2009;37(3):916–30.
Zhao Y-F, Peng T, Sun H-Z, Teotia S, Wen H-L, Du Y-X, et al. miR1432-OsACOT (Acyl-CoA thioesterase) module determines grain yield via enhancing grain filling rate in rice. Plant Biotechnol J. 2019;17(4):712–23.
Yi R, Zhu Z, Hu J, Qian Q, Dai J, Ding Y. Identification and expression analysis of microRNAs at the grain filling stage in rice (Oryza sativa L.) via deep sequencing. PloS one. 2013;8(3):e57863.
Peng T, Sun H, Qiao M, Zhao Y, Du Y, Zhang J, et al. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 2014;14:196.
Luo Z, Zhou Z, Li Y, Tao S, Hu Z-R, Yang J-S, et al. Transcriptome-based gene regulatory network analyses of differential cold tolerance of two tobacco cultivars. BMC Plant Biol. 2022;22(1):369.
Kaur S, Shamshad M, **dal S, Kaur A, Singh S, Sharma A, et al. RNA-seq-based transcriptomics study to investigate the genes governing nitrogen use efficiency in indian wheat cultivars. Front Genet. 2022;13:853910.
Li C, Lai X, Luo K, Zheng Y, Liu K, Wan X. Integrated metabolomic and transcriptomic analyses of two peanut (Arachis hypogaea L.) cultivars differing in amino acid metabolism of the seeds. Plant Physiol Biochem. 2022;185:132–43.
Yuan Z, Dong F, Pang Z, Fallah N, Zhou Y, Li Z, et al. Integrated metabolomics and transcriptome analyses unveil pathways involved in sugar content and rind color of two sugarcane varieties. Front Plant Sci. 2022;13:921536.
Zhang C, Liu H, Zhang H, Dang W, Zhou C, Zhang M. Comparative de novo transcriptome analysis of two cultivars with contrasting content of oil and fatty acids during kernel development in Torreya grandis. Front Plant Sci. 2022;13:909759.
Kumar A, Daware A, Kumar A, Kumar V, Gopala Krishnan S, Mondal S, et al. Genome-wide analysis of polymorphisms identified domestication-associated long low-diversity region carrying important rice grain size/weight quantitative trait loci. Plant J. 2020;103(4):1525–47.
Püffeld M, Seiler C, Kuhlmann M, Sreenivasulu N, Butardo VM Jr. Analysis of develo** rice grain transcriptome using the agilent microarray platform. Methods Mol Biol. 2019;1892:277–300.
Wang GQ, Li HX, Feng L, Chen MX, Meng S, Ye NH, et al. Transcriptomic analysis of grain filling in rice inferior grains under moderate soil drying. J Exp Bot. 2019;70(5):1597–611.
Desai JS, Lawas LMF, Valente AM, Leman AR, Grinevich DO, Jagadish SVK, et al. Warm nights disrupt transcriptome rhythms in field-grown rice panicles. Proc Natl Acad Sci USA. 2021;118(25):e2025899118.
Wang L, Wang D, Yang Z, Jiang S, Qu J, He W, et al. Roles of FERONIA-like receptor genes in regulating grain size and quality in rice. Sci China Life Sci. 2021;64(2):294–310.
**e Q, Xu J, Huang K, Su Y, Tong J, Huang Z, et al. Dynamic formation and transcriptional regulation mediated by phytohormones during chalkiness formation in rice. BMC Plant Biol. 2021;21(1):308.
Li Y, Fan C, **ng Y, Jiang Y, Luo L, Sun L, et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet. 2011;43(12):1266–9.
Wang S, Wu K, Yuan Q, Liu X, Liu Z, Lin X, et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet. 2012;44(8):950–4.
Nallamilli B, Zhang J, Mujahid H, Malone B, Bridges S, Peng Z. Polycomb group gene OsFIE2 regulates rice (Oryza sativa) seed development and grain filling via a mechanism distinct from Arabidopsis. PLoS Genet. 2013;9:e1003322.
Xu H, Bao J-D, Dai J-S, Li Y, Zhu Y. Genome-wide identification of bew reference genes for qRT-PCR normalization under high temperature stress in rice endosperm. PloS one. 2015;10(11):e0142015.
Zhou W, Wang X, Zhou D, Ouyang Y, Yao J. Overexpression of the 16-kDa α-amylase/trypsin inhibitor RAG2 improves grain yield and quality of rice. Plant Biotech J. 2017;15(5):568–80.
Hu Z, Lu S-J, Wang M-J, He H, Sun L, Wang H, et al. A novel QTL qTGW3 encodes the GSK3/SHAGGY-Like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol Plant. 2018;11(5):736–49.
Shi C, Ren Y, Liu L, Wang F, Zhang H, Tian P, et al. Ubiquitin specific protease 15 aas an important role in regulating grain width and size in rice. Plant Physiol. 2019;180(1):381–91.
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–30.
Verma A, Prakash G, Ranjan R, Tyagi AK, Agarwal P. Silencing of an ubiquitin ligase increases grain width and weight in indica rice. Front Genet. 2020;11:600378.
Zhu QH, Spriggs A, Matthew L, Fan L, Kennedy G, Gubler F, et al. A diverse set of microRNAs and microRNA-like small RNAs in develo** rice grains. Genome Res. 2008;18(9):1456–65.
Lacombe S, Nagasaki H, Santi C, Duval D, Piégu B, Bangratz M, et al. Identification of precursor transcripts for 6 novel miRNAs expands the diversity on the genomic organisation and expression of miRNA genes in rice. BMC Plant Biol. 2008;8:123.
Lan Y, Su N, Shen Y, Zhang R, Wu F, Cheng Z, et al. Identification of novel MiRNAs and MiRNA expression profiling during grain development in indica rice. BMC Genom. 2012;13:264.
Yang X, Zhao X, Dai Z, Ma F, Miao X, Shi Z. OsmiR396/growth regulating factor modulate rice grain size through direct regulation of embryo-specific miR408. Plant Physiol. 2021;186(1):519–33.
Showalter AM. Arabinogalactan-proteins: structure, expression and function. Cell Mol Life Sci. 2001;58(10):1399–417.
Lin F, Manisseri C, Fagerström A, Peck ML, Vega-Sánchez ME, Williams B, et al. Cell wall composition and candidate biosynthesis gene expression during rice development. Plant Cell Physiol. 2016;57(10):2058–75.
Kondou Y, Nakazawa M, Kawashima M, Ichikawa T, Yoshizumi T, Suzuki K, et al. RETARDED GROWTH OF EMBRYO1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol. 2008;147(4):1924–35.
Huang L-M, Lai C-P, Chen L-F, Chan M-T, Shaw J-F. Arabidopsis SFAR4 is a novel GDSL-type esterase involved in fatty acid degradation and glucose tolerance. Bot Stud. 2015;56:33.
Rosario A, Briones V, Vidal A, Juliano B. Composition and endosperm structure of develo** and mature rice kernel. Cereal Chem. 1968;45:225–35.
Roberts JK, DeSimone NA, Lingle WL, Dure L 3rd. Cellular concentrations and uniformity of cell-type accumulation of two LEA proteins in cotton embryos. Plant Cell. 1993;5(7):769–80.
Hong-Bo S, Zong-Suo L, Ming-An S. LEA proteins in higher plants: structure, function, gene expression and regulation. Colloids Surf B. 2005;45(3–4):131–5.
Kowles RV, Phillips RL. DNA amplification patterns in maize endosperm nuclei during kernel development. Proc Natl Acad Sci. 1985;82(20):7010–4.
Kladnik A, Chourey PS, Pring DR, Dermastia M. Development of the endosperm of Sorghum bicolor during the endoreduplication-associated growth phase. J Cereal Sci. 2006;43(2):209–15.
Melaragno JE, Mehrotra B, Coleman AW. Relationship between endopolyploidy and cell size in epidermal tissue of Arabidopsis. Plant Cell. 1993;5(11):1661–8.
Larkins BA, Dilkes BP, Dante RA, Coelho CM, Woo YM, Liu Y. Investigating the hows and whys of DNA endoreduplication. J Exp Bot. 2001;52(355):183–92.
Sugimoto-Shirasu K, Roberts K. “Big it up”: endoreduplication and cell-size control in plants. Curr Opin Plant Biol. 2003;6(6):544–53.
Shaw BP, Sekhar S, Panda BB, Sahu G, Chandra T, Parida AK. Biochemical and molecular processes contributing to grain filling and yield in rice. Plant Physiol Biochem. 2022;179:120–33.
He W, Wang L, Lin Q, Yu F. Rice seed storage proteins: biosynthetic pathways and the effects of environmental factors. J Integr Plant Biol. 2021;63(12):1999–2019.
Tan X, Tan X, Li E, Bai Y, Nguyen T. Starch molecular fine structure is associated with protein composition in chickpea seed. Carbohydr Polym. 2021;272:118489.
** M, Wu W, Xu Y, Zhou Y, Chen G, Ji Y, et al. Grain chalkiness traits is affected by the synthesis and dynamic accumulation of the storage protein in rice. J Sci Food Agric. 2021;101(14):6125–33.
Zhang L, Zhang C, Yan Y, Hu Z, Wang K, Zhou J, et al. Influence of starch fine structure and storage proteins on the eating quality of rice varieties with similar amylose contents. J Sci Food Agric. 2021;101(9):3811–8.
Matsuda T. Rice flour: a promising food material for nutrition and global health. J Nutr Sci Vitaminol. 2019;65(Supplement):S13–7.
Gibbings JG, Cook BP, Dufault MR, Madden SL, Khuri S, Turnbull CJ, et al. Global transcript analysis of rice leaf and seed using SAGE technology. Plant Biotechnol J. 2003;1(4):271–85.
Geigenberger P, Stitt M. Sucrose synthase catalyses a readily reversible reaction in vivo in develo** potato tubers and other plant tissues. Planta. 1993;189(3):329–39.
Amor Y, Haigler CH, Johnson S, Wainscott M, Delmer DP. A membrane-associated form of sucrose synthase and its potential role in synthesis of cellulose and callose in plants. Proc Natl Acad Sci USA. 1995;92(20):9353–7.
Harada T, Satoh S, Yoshioka T, Ishizawa K. Expression of sucrose synthase genes involved in enhanced elongation of pondweed (Potamogeton distinctus) turions under anoxia. Ann Bot. 2005;96(4):683–92.
Jiang H, Dian W, Liu F, Wu P. Isolation and characterization of two fructokinase cDNA clones from rice. Phytochemistry. 2003;62(1):47–52.
Satoh H, Shibahara K, Tokunaga T, Nishi A, Tasaki M, Hwang S-K, et al. Mutation of the plastidial α-GLUCAN PHOSPHORYLASE gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell. 2008;20(7):1833–49.
Hori K, Sun J. Rice grain size and quality. Rice. 2022;15(1):33.
Guo X, Fu Y, Lee Y-RJ, Chern M, Li M, Cheng M, et al. The PGS1 basic helix-loop-helix protein regulates Fl3 to impact seed growth and grain yield in cereals. Plant Biotechnol J. 2022;20(7):1311–26.
Cai Y, Chen H, **ao N, Wu Y, Yu L, Chen Z, et al. Substandard starch grain4 may function in amyloplast development by influencing starch and lipid metabolism in rice endosperm. J Plant Physiol. 2022;270:153638.
Huang M, Chen J, Cao F, Tao Z, Lei T, Tian A, et al. Quantifying accumulation characteristics of glutelin and prolamin in rice grains. PLoS ONE. 2019;14(7):e0220139.
**ong Y, Ren Y, Li W, Wu F, Yang W, Huang X, et al. NF-YC12 is a key multi-functional regulator of accumulation of seed storage substances in rice. J Exp Bot. 2019;70(15):3765–80.
Chattopadhyay K, Behera L, Bagchi TB, Sardar SS, Moharana N, Patra NR, et al. Detection of stable QTLs for grain protein content in rice (Oryza sativa L.) employing high throughput phenoty** and genoty** platforms. Sci Rep. 2019;9(1):3196.
Wang JC, Xu H, Zhu Y, Liu QQ, Cai XL. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J Exp Bot. 2013;64(11):3453–66.
Kumar P, Mishra A, Sharma H, Sharma D, Rahim MS, Sharma M, et al. Pivotal role of bZIPs in amylose biosynthesis by genome survey and transcriptome analysis in wheat (Triticum aestivum L.) mutants. Sci Rep. 2018;8(1):17240.
Xu H, Li S, Kazeem BB, Ajadi AA, Luo J, Yin M, et al. Five rice seed-specific NF-YC genes redundantly regulate grain quality and seed germination via interfering gibberellin pathway. Int J Mol Sci. 2022;23(15):8382.
E Z, Li T, Zhang H, Liu Z, Deng H, Sharma S, et al. A group of nuclear factor Y transcription factors are sub-functionalized during endosperm development in monocots. J Exp Bot. 2018;69(10):2495–510.
Bai AN, Lu XD, Li DQ, Liu JX, Liu CM. NF-YB1-regulated expression of sucrose transporters in aleurone facilitates sugar loading to rice endosperm. Cell Res. 2016;26(3):384–8.
Feng T, Wang L, Li L, Liu Y, Chong K, Theißen G, et al. OsMADS14 and NF-YB1 cooperate in the direct activation of OsAGPL2 and WAXY during starch synthesis in rice endosperm. New Phytol. 2022;234(1):77–92.
Yin L-L, Xue H-W. The MADS29 transcription factor regulates the degradation of the nucellus and the nucellar projection during rice seed development. Plant Cell. 2012;24(3):1049–65.
Zhao G, **e S, Zong S, Wang T, Mao C, Shi J, et al. Mutation of TL1, encoding a novel C(2)H(2) zinc finger protein, improves grains eating and cooking quality in rice. Theor Appl Genet. 2022;135(10):3531–43.
Li J, Lee GI, Van Doren SR, Walker JC. The FHA domain mediates phosphoprotein interactions. J Cell Sci. 2000;113(Pt 23):4143–9.
Bochar DA, Savard J, Wang W, Lafleur DW, Moore P, Côté J, et al. A family of chromatin remodeling factors related to williams syndrome transcription factor. Proc Natl Acad Sci. 2000;97(3):1038–43.
Loewith R, Meijer M, Lees-Miller SP, Riabowol K, Young D. Three yeast proteins related to the human candidate tumor suppressor p33(ING1) are associated with histone acetyltransferase activities. Mol Cell Biol. 2000;20(11):3807–16.
Sekimata M, Homma Y. Regulation of Rb gene expression by an MBD2-interacting zinc finger protein MIZF during myogenic differentiation. Biochem Biophys Res Commun. 2004;325(3):653–9.
Lu C-A, Ho T-HD, Ho S-L, Yu S-M. Three novel MYB proteins with one DNA binding repeat mediate sugar and hormone regulation of α-amylase gene expression. Plant Cell. 2002;14(8):1963–80.
Lv Y, Yang M, Hu D, Yang Z, Ma S, Li X, et al. The OsMYB30 transcription factor suppresses cold tolerance by interacting with a JAZ Protein and suppressing β-amylase expression. Plant Physiol. 2017;173(2):1475–91.
Kaneda T, Taga Y, Takai R, Iwano M, Matsui H, Takayama S, et al. The transcription factor OsNAC4 is a key positive regulator of plant hypersensitive cell death. EMBO J. 2009;28(7):926–36.
Zhong R, Lee C, McCarthy RL, Reeves CK, Jones EG, Ye Z-H. Transcriptional activation of secondary wall biosynthesis by rice and maize NAC and MYB transcription factors. Plant Cell Physiol. 2011;52(10):1856–71.
Gao F, **ong A, Peng R, ** X, Xu J, Zhu B, et al. OsNAC52, a rice NAC transcription factor, potentially responds to ABA and confers drought tolerance in transgenic plants. Plant Cell Tissue Organ Cult. 2010;100(3):255–62.
Liu J, Wu MW, Liu CM. Cereal endosperms: development and storage product accumulation. Annu Rev Plant Biol. 2022;73:255–91.
Kant S, Bi YM, Zhu T, Rothstein SJ. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009;151(2):691–701.
McAdam EL, Meitzel T, Quittenden LJ, Davidson SE, Dalmais M, Bendahmane AI, et al. Evidence that auxin is required for normal seed size and starch synthesis in pea. New Phytol. 2017;216(1):193–204.
Qiao J, Jiang H, Lin Y, Shang L, Wang M, Li D, et al. A novel miR167a-OsARF6-OsAUX3 module regulates grain length and weight in rice. Mol Plant. 2021;14(10):1683–98.
Cheng ZJ, Zhao XY, Shao XX, Wang F, Zhou C, Liu YG, et al. Abscisic acid regulates early seed development in Arabidopsis by ABI5-mediated transcription of SHORT HYPOCOTYL UNDER BLUE1. Plant Cell. 2014;26(3):1053–68.
Ashikari M, Wu J, Yano M, Sasaki T, Yoshimura A. Rice gibberellin-insensitive dwarf mutant gene DWARF 1 encodes the alpha-subunit of GTP-binding protein. Proc Natl Acad Sci USA. 1999;96(18):10284–9.
Tong H, **ao Y, Liu D, Gao S, Liu L, Yin Y, et al. Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell. 2014;26(11):4376–93.
Wang L, Wang Z, Xu Y, Joo SH, Kim SK, Xue Z, et al. OsGSR1 is involved in crosstalk between gibberellins and brassinosteroids in rice. Plant J. 2009;57(3):498–510.
Sakamoto T, Morinaka Y, Ishiyama K, Kobayashi M, Itoh H, Kayano T, et al. Genetic manipulation of gibberellin metabolism in transgenic rice. Nat Biotechnol. 2003;21(8):909–13.
Yang J, Zhang J, Wang Z, Liu K, Wang P. Post-anthesis development of inferior and superior spikelets in rice in relation to abscisic acid and ethylene. J Exp Bot. 2006;57(1):149–60.
Young TE, Gallie DR, DeMason DA. Ethylene-mediated programmed cell death during maize endosperm development of wild-type and SHRUNKEN2 Genotypes. Plant Physiol. 1997;115(2):737–51.
Peng T, Lv Q, Zhang J, Li J, Du Y, Zhao Q. Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J Exp Bot. 2011;62(14):4943–54.
Holland BL, Monk NAM, Clayton RH, Osborne CP. A theoretical analysis of how plant growth is limited by carbon allocation strategies and respiration. In Silico Plants. 2019;1(1):diz004.
**a K, Ou X, Tang H, Wang R, Wu P, Jia Y, et al. Rice microRNA osa-miR1848 targets the obtusifoliol 14α-demethylase gene OsCYP51G3 and mediates the biosynthesis of phytosterols and brassinosteroids during development and in response to stress. New Phytol. 2015;208(3):790–802.
Zhang YC, Yu Y, Wang CY, Li ZY, Liu Q, Xu J, et al. Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat Biotechnol. 2013;31(9):848–52.
Du P, Wu J, Zhang J, Zhao S, Zheng H, Gao G, et al. Viral infection induces expression of novel phased microRNAs from conserved cellular microRNA precursors. PLoS Pathog. 2011;7:e1002176.
Mangrauthia SK, Bhogireddy S, Agarwal S, Prasanth VV, Voleti SR, Neelamraju S, et al. Genome-wide changes in microRNA expression during short and prolonged heat stress and recovery in contrasting rice cultivars. J Exp Bot. 2017;68(9):2399–412.
Agarwal P, Singh PC, Chaudhry V, Shirke PA, Chakrabarty D, Farooqui A, et al. PGPR-induced OsASR6 improves plant growth and yield by altering root auxin sensitivity and the xylem structure in transgenic Arabidopsis thaliana. J Plant Physiol. 2019;240:153010.
Li Y, Zheng Y-P, Zhou X-H, Yang X-M, He X-R, Feng Q, et al. Rice miR1432 fine-tunes the balance of yield and blast disease resistance via different modules. Rice. 2021;14(1):87.
Duan P, Ni S, Wang J, Zhang B, Xu R, Wang Y, et al. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat Plants. 2015;2:15203.
Yan Y, Wei M, Li Y, Tao H, Wu H, Chen Z, et al. MiR529a controls plant height, tiller number, panicle architecture and grain size by regulating SPL target genes in rice (Oryza sativa L.). Plant Sci. 2021;302:110728.
Iwamoto M, Tagiri A. MicroRNA-targeted transcription factor gene RDD1 promotes nutrient ion uptake and accumulation in rice. Plant J. 2016;85(4):466–77.
Zhang H, Zhang J, Yan J, Gou F, Mao Y, Tang G, et al. Short tandem target mimic rice lines uncover functions of miRNAs in regulating important agronomic traits. Proc Natl Acad Sci USA. 2017;114(20):5277–82.
Marowa P, Ding A, Kong Y. Expansins: roles in plant growth and potential applications in crop improvement. Plant Cell Rep. 2016;35(5):949–65.
Li S, Gao F, **e K, Zeng X, Cao Y, Zeng J, et al. The OsmiR396c-OsGRF4-OsGIF1 regulatory module determines grain size and yield in rice. Plant Biotechnol J. 2016;14(11):2134–46.
Miao C, Wang D, He R, Liu S, Zhu J-K. Mutations in MIR396e and MIR396f increase grain size and modulate shoot architecture in rice. Plant Biotechnol J. 2020;18(2):491–501.
Gao F, Wang K, Liu Y, Chen Y, Chen P, Shi Z, et al. Blocking miR396 increases rice yield by sha** inflorescence architecture. Nat Plants. 2015;2(1):15196.
Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development. 2010;137(1):103–12.
Qin R, Zeng D, Yang C, Akhter D, Alamin M, ** X, et al. LTBSG1, a new allele of BRD2, regulates panicle and grain development in rice by brassinosteroid biosynthetic pathway. Genes. 2018;9(6):292.
Shen L, Zhang Q, Wang Z, Wen H, Hu G, Ren D, et al. OsCAF2 contains two CRM domains and is necessary for chloroplast development in rice. BMC Plant Biol. 2020;20(1):381.
Li Z, An X, Zhu T, Yan T, Wu S, Tian Y, et al. Discovering and constructing ceRNA-miRNA-target gene regulatory networks during anther development in maize. Int J Mol Sci. 2019;20(14):3480.
Guo W, Wu G, Yan F, Lu Y, Zheng H, Lin L, et al. Identification of novel Oryza sativa miRNAs in deep sequencing-based small RNA libraries of rice infected with rice stripe virus. PLoS ONE. 2012;7(10):e46443.
Liu H, Wang R, Mao B, Zhao B, Wang J. Identification of lncRNAs involved in rice ovule development and female gametophyte abortion by genome-wide screening and functional analysis. BMC Genom. 2019;20(1):90.
Zhao J, Liu X, Wang M, **e L, Wu Z, Yu J, et al. The miR528-D3 module regulates plant height in rice by modulating the gibberellin and abscisic acid metabolisms. Rice. 2022;15(1):27.
Zhang YC, He RR, Lian JP, Zhou YF, Zhang F, Li QF, et al. OsmiR528 regulates rice-pollen intine formation by targeting an uclacyanin to influence flavonoid metabolism. Proc Natl Acad Sci USA. 2020;117(1):727–32.
Yang R, Li P, Mei H, Wang D, Sun J, Yang C, et al. Fine-tuning of miR528 accumulation modulates flowering time in rice. Mol Plant. 2019;12(8):1103–13.
Fang N, Xu R, Huang L, Zhang B, Duan P, Li N, et al. SMALL GRAIN 11 controls grain size, grain number and grain yield in rice. Rice. 2016;9(1):64.
Bae KD, Um TY, Yang WT, Park TH, Hong SY, Kim KM, et al. Characterization of dwarf and narrow leaf (dnl-4) mutant in rice. Plant Signal Behav. 2021;16(2):1849490.
Zhan P, Wei X, **ao Z, Wang X, Ma S, Lin S, et al. GW10, a member of P450 subfamily regulates grain size and grain number in rice. Theor Appl Genet. 2021;134(12):3941–50.
Ichihara KI, Kobayashi N, Saito K. Lipid synthesis and acyl-CoA synthetase in develo** rice seeds. Lipids. 2003;38(8):881–4.
Ding LN, Gu SL, Zhu FG, Ma ZY, Li J, Li M, et al. Long-chain acyl-CoA synthetase 2 is involved in seed oil production in Brassica napus. BMC Plant Biol. 2020;20(1):21.
Takahashi K, Morimoto R, Tabeta H, Asaoka M, Ishida M, Maeshima M, et al. Compensated cell enlargement in fugu5 is specifically triggered by lowered sucrose production from seed storage lipids. Plant Cell Physiol. 2017;58(4):668–78.
Araki S, Le NT, Koizumi K, Villar-Briones A, Nonomura KI, Endo M, et al. miR2118-dependent U-rich phasiRNA production in rice anther wall development. Nat Commun. 2020;11(1):3115.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Singh G, Kumar S, Singh P. A quick method to isolate RNA from wheat and other carbohydrate-rich seeds. Plant Mol Biol Rep. 2003;21(1):93-.
Mathew IE, Das S, Mahto A, Agarwal P. Three rice NAC transcription factors heteromerize and are associated with seed size. Front Plant Sci. 2016;7:1638.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78.
Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38(Web Server issue):W64-70.
Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–8.
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37(6):914–39.
Wickham H. Resha** data with the reshape Package. J Stat Softw. 2007;21:1–20.
Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010. Chapter 11:Unit 11.7.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Dai X, Zhuang Z, Zhao PX. psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018;46(W1):W49-w54.
Cakir B, Tian L, Crofts N, Chou H-L, Koper K, Ng C-Y, et al. Re-programming of gene expression in the CS8 rice line over-expressing ADP glucose pyrophosphorylase induces a suppressor of starch biosynthesis. Plant J. 2019;97(6):1073–88.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Acknowledgements
A.M., A.Y., and A.P.V. acknowledge University Grants Commission for Ph. D. research fellowship; P.A. is thankful to the Department of Biotechnology, and Department of Science and Technology, Ministry of Science and Technology, India, for grants supporting the research in this paper; and to NIPGR for core grant. The authors acknowledge the support provided by central instrumentation and microscopy facilities of NIPGR and DBT e-library consortium for providing online access to articles.
The authors are grateful to StatWorld Research Solutions Pvt. Ltd. (https://www.statworld.in/) for generating the database.
The authors acknowledge Priya Jaiswal and Richa Priyadarshini for their inputs.
The authors dedicate this article to the memory of Prof. Jitendra P. Khurana, Department of Plant Molecular Biology, University of Delhi, South Campus, New Delhi, who had given his valuable scientific suggestions for the manuscript and was also an excellent mentor to P.A.
Funding
This work was supported by research grant numbers BT/AB/NIPGR/SEED BIOLOGY/2012 by Department of Biotechnology (DBT) and SB/WEA-02/2014, CRG/2018/000501 and SPG/2021/002899 by Science and Engineering Research Board (SERB) under Department of Science and Technology (DST); both under the Ministry of Science and Technology, India; and by National Institute of Plant Genome Research (NIPGR) core grant to PA.
Author information
Authors and Affiliations
Contributions
A.M. performed all the experiments. A.Y. and A.P.V. performed the validation experiments. S.K.P. provided the QTL data. A.M. and P.A. wrote the manuscript. A.K.T. and P.A. formulated and supervised the study. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
(related to Fig. 1). Morphological changes in seeds of SN and LGR during development. (A) SEM images of husk of SN (left) and LGR (right) showing the middle portion of lemma. Individual cells have been marked by asterisks (scale bar = 100μm). (B) Images of seeds of SN and LGR. S1–S5 represent five stages of rice seed development. Left and right panels contain seeds (husked and dehusked) of SN and LGR from representative DAP of the five seed stages. (C) An additional set of endosperm sections of SN (left) and LGR (right) at DAP mentioned on the left side, representing S2, S3 and S4 stages of seed development stained with toluidine blue-O. i, v, ix, xiii, xvii and iii, vii, xi, xv, xix show central endosperm of SN and LGR, respectively, while ii, vi, x, xiv, xviii and iv, viii, xii, xvi, xx show peripheral endosperms of SN and LGR, respectively. Red and blue triangles indicate nuclei and cell wall, respectively. Scale bar = 50 µm. Figure S2. Correlation between biological replicates of SN and LGR. (A) Validation of RNA sequencing data by qPCR of five genes in SN and LGR. In each box (gene names are mentioned at the top), the upper panel indicates SN and lower panel indicates LGR. The light brown and light green bars represent relative expression (log2fold change) values from RNA sequencing (n=3; q value≤0.05) and the dark brown and dark green bars represent relative qPCR expression values (n=2; error bars represent ± SD), respectively. S1–S5 represent five stages of seed development and Leaf represents flag leaf. See replicate data in Additional file 2: Table S18. (B) Pearson’s correlation between the three biological replicates (_1, _2, _3) from five seed stages (S1–S5) and flag leaf (Leaf) of SN (top) and LGR (bottom). Color legend represents the value of Pearson’s correlation coefficient ranging from -1 to +1. Blue color represents high correlation. (C) PCA plot representing grou** patterns of biological replicates. The plot depicts the relatedness between the biological replicates of a sample as well as between the seed stages and leaf tissue in terms of their transcriptome. The color coding used for denoting the samples in the plot has been shown in the color legend. Figure S3. Expression profiles of SN and LGR. The Venn diagram represents the total number of genes expressed specifically in seeds of SN and LGR, i.e., these genes are not expressed (FPKM<1) in the flag leaf. GO enrichment analysis of seed-specific genes common between SN and LGR (as marked in red in the figure) was performed using BINGO plug-in of Cytoscape. Purple and violet colors indicate enriched GO categories at p values mentioned in the color legend. White color indicates no significant enrichment. Highly enriched GO terms have been encircled. Interaction between seed-specific GO terms has been shown by means of edges (arrows) in the network diagram. Nodes = GO terms, edges = interactions. Figure S4. Analysis of differential expression profiles of seeds of SN and LGR. (A) Total number of differentially expressed genes obtained in seed developmental stages of SN and LGR. The genes were filtered at the cutoffs of FPKM≥1, log2fc≥1 and q value≤0.05. Violet and green bars indicate SN and LGR, respectively. (B) Genes with log2fc≥10 in SN and LGR were annotated using MapMan (violet and green bars indicate SN and LGR, respectively as indicated in the color legend; bars on the right and left sides represent up- and downregulated genes, respectively). Pathways with ≥50 genes upregulated in both SN and LGR have been marked with red arrows. Pathways with significant number of genes differentially expressed only in SN and LGR have been marked with asterisks (blue = SN, pink = LGR). (C) Transcription factor families with ≥5 upregulated members with log2fc≥10 in both SN and LGR have been plotted (solid blue bars represent SN and bars with pattern filling represent LGR as shown in the color legend). Figure S5. Comparison of stage-specific DEGs. Stage-specific DEGs (log2fc≥1 in one stage and log2fc<1 in rest of the four stages in a genotype) were compared to study the similarity in the transcriptomes of SN and LGR. Bars above and below the X-axis represent up and down regulation, respectively, as indicated. Figure S6. (related to Fig. 3). Differential expression analysis of genes related to cell cycle in SN and LGR. (A) GO enrichment analysis of all upregulated genes of SN and LGR, respectively, was performed by AgriGO. Yellow, orange and red colors indicate significant enrichment as denoted in the color legend. The relationship between GO terms is as indicated in the color legend by arrows. GO terms enriched specifically and enriched at higher p-value in LGR have been demarcated with red and blue dotted boxes, respectively. (B) The distribution of the cell cycle genes specifically upregulated in SN (denoted by the black box) was studied in the seed developmental stages of SN (as represented in the color legend). S5 stage of SN had the maximum number of cell cycle related DEGs (marked by asterisks in the graph). (C) FPKM values of genes, which promote endoreduplication, and are commonly upregulated in SN and LGR but exhibit different expression patterns (such as variation in peak expression) between the two genotypes. These included genes encoding Wee1 kinases and mitotic spindle checkpoint protein MAD2, which inhibit anaphase progression. Figure S7. (related to Fig. 3). Functional annotation of LGR S3-preferential DEGs. (A) Per cent similarity between DEGs of consecutive stages of SN (bars above and below the axis represent up- and downregulated genes, respectively). (B) Pathway analysis of DEGs (by MapMan) that are LGR S3 preferential, i.e up regulated in LGR S3 but not in LGR S2 and SN S2. Genes related to degradation of starch, TCA cycle, mitochondrial ETC and cell wall modification were upregulated (demarcated with pink boxes in the figure). Figure S8. (related to Fig. 4). Expression pattern of carbohydrate and PCD-related genes in seeds of SN and LGR. (A) A phosphofructokinase and an α-glucan phosphorylase gene showed high expression levels (log2FPKM≥5) in the seed tissues of SN and LGR. Arrows indicate peak in expression values (log2FPKM). (B) Heatmap representing log2FPKM values of known genes related to PCD in seed and leaf tissues of SN and LGR (color legend at the bottom). The positive and negative regulators of PCD showing difference in expression levels and pattern between SN and LGR have been marked in red and green boxes, respectively. (C) Graph showing log2FPKM values of four positive and one negative regulator of PCD, marked in (B). Positive and negative regulators have been denoted by round and square markers, respectively. Figure S9. (related to Fig. 6). Categorization of differentially expressed TFs based on their regulation patterns amongst SN and LGR. TFs that were DE (815 and 545 upregulated while 545 and 480 downregulated in SN and LGR, respectively) in SN and LGR seeds were divided into four categories [up in any stage in both SN and LGR (473 TFs; from 59 families), down in any stage in both SN and LGR (340 TFs; from 54 families), up in any stage in SN and down in LGR (102 TFs; 30 families), up in any stage in LGR and down in SN (28 TFs; 18 families); pie chart in the center] based on their regulation. Left and right panels indicate TF families with similar and opposite regulation pattern, respectively. Prominent TF families (with ≥20 members) showing similar regulation pattern in SN and LGR have been marked with asterisks in the color legend (number of members has been written on the pie charts). Figure S10. (related to Fig. 7). Expression analysis of hormone signaling genes with different expression patterns in SN and LGR. (A-H) Heat maps showing expression levels (log2FPKM) of genes related to auxin, ABA, GA, ethylene, BR, CK, JA and SA signaling, respectively, that showed different expression pattern between SN and LGR, and hence, were present in separate co-expression clusters. In each heat map, the upper and lower panels show expression levels in five stages of seed development (S1–S5) and flag leaf (Leaf) in SN and LGR, respectively, as mentioned along with the locus ID of the gene. Red dots indicate genes that have higher expression (log2FPKM≥0.5) in any stage in either genotype, and have been plotted in Fig. 7. SY = synthesis, DG = degradation, SG = signal transduction, RS = response. Figure S11. Expression analysis of genes reported for regulating seed size and weight in rice. (A, B) Regulation pattern of genes reported in literature in seed developmental stages of SN and LGR as mentioned in the figure. (C) Heat map based on log2FPKM values and patterns of genes regulating seed size and weight in the seed developmental stages (S1–S5) and leaf tissues of SN and LGR. Red dot = negative regulator of seed size and weight, green dot = positive regulator of seed size and weight, blue dot = affects both seed length and width. The arrows indicate comparison of expression patterns and levels between SN and LGR. Genes with similar expression pattern between SN and LGR have been marked with an orange or black arrow. In this, dark orange arrow marks genes with higher expression level (Log2FPKM≥1) in SN while light orange arrow marks the same genes with lower expression in LGR. Gray arrow marks genes with higher expression (Log2FPKM≥1) in LGR while black arrow marks the same genes with lower expression level in SN. Since these genes have similar expression pattern, the homologs from SN and LGR mostly appear close to each other in the hierarchy. Green arrows mark genes with different expression pattern between SN and LGR, with dark and light green indicating SN and LGR, respectively. Blue box is for genes which do not express in both SN and LGR (as indicated by color legend on top). (D) Graphs representing expression (FPKM) of genes known to regulate seed size and weight in rice in the five seed stages of SN and LGR. Graphs a-h show genes with different expression patterns between SN and LGR; graphs i-m show genes with similar expression patterns but different expression levels amongst SN and LGR. S1–S5 represent five seed stages, locus IDs and gene names have been mentioned above each graph, brown and green lines represent SN and LGR, respectively. Figure S12. Expression profiles of miRNAs in the seeds of SN and LGR. (A) Validation of sRNAseq data by stem-loop qRT-PCR of miR530-5pin SN (left panel) and LGR (right panel) (n=3). The lower panel represents log2 fold change by sRNAseq (See replicate data in Additional file 2: Table S18). (B) Total number of miRNAs expressed (TPM≥50) in seed developmental stages of SN and LGR (ovals on top) and corresponding number of families (ovals at bottom). The number of families specific (miRNAs with TPM≥50 in one genotype and TPM<50 in the other) to and common (TPM≥50 in both genotypes) between SN and LGR have been written in black and red, respectively. Families with miRNAs expressed specifically in SN and LGR have been mentioned below the Venn diagram. (C) Number of miRNAs that are specific to (miRNAs with TPM≥50 in one stage and TPM<50 in the other stages) and common (TPM≥50 in all five stages) between seed developmental stages (S1–S5) of SN and LGR. Green and orange bars represent specific miRNAs in SN and LGR, respectively (as indicated in the color legend). Blue ovals represent miRNAs common between SN and LGR. Figure S13. Differential expression profile of miRNAs in the seeds of SN and LGR. (A) Total number of differentially expressed miRNAs (DEMs, log2fc≥2) obtained in seed developmental stages of SN and LGR with respect to flag leaf. Blue bars represent SN and orange bars represent LGR (as indicated in the color legend). (B) Comparison of DEMs between same stages/SS and comparable stages/CS of SN and LGR. Numbers of DEMs for SS are in blue while for CS are in brown. Figure S14. miRNAs commonly differentially expressed in all five stages of SN or LGR and their target mRNAs. Graphs show differential expression levels (log2fc) of miRNAs (A) up- and (B) downregulated in all five stages of each genotype. The targets have negative correlation in expression and are also down or upregulated, respectively, in all stages of the genotype. For both A and B, upper panel is for SN while lower one is for LGR. In A, bars above and below the X axis represent miRNAs up- and their targets downregulated, respectively. In B, bars above and below the X axis represent miRNAs down- and their targets upregulated, respectively. The horizontal trendlines in each graph depict negative correlation amongst differential expression of miRNAs and their targets. Figure S15. Functional overlay of whole transcriptome data. Functional annotation of genes derived from seven analyses (conditions) of whole transcriptome sequencing to find out genes involved in/coding for TFs, hormone signaling, cell cycle, cell growth, SSPs and carbohydrate metabolism with the overall analyses. The description of the analyses is given in the first column. Description of the genotype and stage is given in the next column. The total numbers of genes present in that particular category, which were used for subsequent functional annotation, have been mentioned in parenthesis. For each analysis, the numbers of genes falling in different functional categories have been mention in respective rows. Figure S16. Screenshot of Rice Grain Development Database (RGDD). (A) The transcriptome tab allows search either by locus ID (option 1) or by function (option 2). The functions enlisted are transcription factors and phytohormones (related genes). Transposable elements can also be selected for. A gene list is displayed for option 2, from which locus IDs can be selected. Both expression and log2 fold change data are shown as graphs. (B) The miRNome tab allows for search as miRNA ID (option 1) or from a list of expressed miRNAs (option 2). Both expression and log2 fold change data are displayed as line graphs.
Additional file 2: Table S1.
Summary of reads obtained from total RNA sequencing of 36 cDNA libraries from the five seed developmental stages (S1-S5) and flag leaf (L) of SN and LGR. Table S2. Pearson’s correlation between biological replicates of the five seed developmental stages (S1-S5) and flag leaf (L) of SN and LGR. Table S3. FPKM values of all rice genes (transposable elements removed) in flag leaf and five seed development tissues of SN and LGR. The higlighted ones indicate significant DEGs in at least one seed stage. Table S4. Log2fold change values in seed development stages, of significant DEGs (Log2FC≥1, q value≤0.05, p value ≤0.05, FPKM≥1). Non-significant differential expression is indicated by ‘-’. Table S5. Stage-specific DEGs common between SN and LGR. Log2 fold change values of DEGs which are specific (log2FC≥1 in that stage and <1 in rest) to the same stage of SN and LGR. Table S6. FPKM values of cell cycle related genes specifically up regulated in seeds of SN and LGR. Table S7. List of DEGs preferential to S3 stage of LGR by comparison with SN S3/LGR S2 and SN S2/LGR S2. Genes which are LGR S3 preferential in both analyses have been highlighted. Table S8. List of TF encoding genes (with their log2fold changes), significantly differentially expressed at log2fc≥1 in at least one seed developmental stage of SN and/or LGR. “-” indicates non-significant values. Table S9. FPKM values of phytohormone encoding genes, which were present in different expression clusters (variation in expression pattern) and had higher expression values (difference in log2FPKM≥0.5; variation in expression level) in a stage in either SN or LGR. Table S10. Summary of grain size QTLs identified using a map** population (Sonasal x LGR) in rice. Table S11. Log2 fold change values of genes which are significantly differentially expressed amongst SN and LGR seed development stages and are located within QTLs associated with a seed size-related trait. Table S12a. Summary of reads obtained from small RNA sequencing of each library from the five seed developmental stages (S1-S5) and flag leaf (L) of SN. In all samples, _1, _2, _3 represent biological replicates. Table S12b. Summary of reads obtained from small RNA sequencing of each library from the five seed developmental stages (S1-S5) and flag leaf (L) of LGR. In all samples, _1, _2, _3 represent biological replicates. Table S13. Pearson's correlation between biological replicates of the five seed developmental stages (S1-S5) and flag leaf (L) of SN and LGR obtained from small RNA sequencing. Table S14. Expression of all miRs in seed and flag leaf of SN and LGR. The TPM values of 604 miRs detected in at least one of the tissues used has been mentioned. The TPM values ≥50 have been highlighted in gray. Table S15. Comparison of DEMs between SN and LGR. The orange colored cells indicate the log2 fold changes of 467 DEMs for each stage of seed development. The yellow color indicates comparison between similar stages (by developmental event) of SN and LGR. The grey color indicates comparison between same stage (by DAP) of SN and LGR. U implies 'up regulation' while D implies 'down regulation' in the respective stage mentioned. - indicates non-significant differential expression. The up regulated genes have been marked in green while down regulated ones are in red. Table S16. List of miRNA-target modules for miRNAs up and down-regulated in all five seed developmental stages of either SN or LGR. Table S17. List of primers for validation. Table S18. Details of replicate data.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mahto, A., Yadav, A., P. V., A. et al. Cytological, transcriptome and miRNome temporal landscapes decode enhancement of rice grain size. BMC Biol 21, 91 (2023). https://doi.org/10.1186/s12915-023-01577-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-023-01577-3