
Abstract
Background
Oxytocin, secreted by oxytocin neurons in the hypothalamus, is an endogenous neuropeptide involved in modulating multiple sensory information processing pathways, and its roles in the brain have been associated with prosocial, maternal, and feeding-related behaviors. Visual information is necessary for initiating these behaviors, with the retina consisting of the first stage in the visual system mediating external stimulus perception. Oxytocin has been detected in the mammalian retina; however, the expression and possible function of oxytocin receptors (OxtR) in the retina remain unknown. Here, we explore the role of oxytocin in regulating visual information processing in the retina.
Results
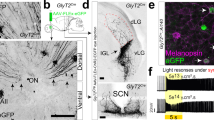
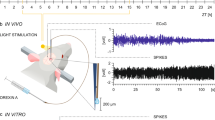
We observed that OxtR mRNA and protein are expressed in the mouse retina. With Oxtr-Cre transgenic mice, immunostaining, and fluorescence in situ hybridization, we found that OxtRs are mainly expressed in GABAergic amacrine cells (ACs) in both the inner nuclear layer (INL) and ganglion cell layer (GCL). Further immunoreactivity studies showed that GABAergic OxtR+ neurons are mainly cholinergic and dopaminergic neurons in the INL and are cholinergic and corticotrophin-releasing hormone neurons in the GCL. Surprisingly, a high level of Oxtr mRNAs was detected in retinal dopaminergic neurons, and exogenous oxytocin application activated dopaminergic neurons to elevate the retinal dopamine level. Relying on in vivo electroretinographic recording, we found that activating retinal OxtRs reduced the activity of bipolar cells via OxtRs and dopamine receptors.
Conclusions
These data indicate the functional expression of OxtRs in retinal GABAergic ACs, especially dopaminergic ACs, and expand the interactions between oxytocinergic and dopaminergic systems. This study suggests that visual perception, from the first stage of information processing in the retina, is modulated by hypothalamic oxytocin signaling.
Similar content being viewed by others

Background
In the vertebrate visual system, the retina initiates visual information processing. While retinal neurons use synaptic neurotransmitters, including glutamate, gamma-aminobutyric acid (GABA), and glycine, to encode and transmit visual information, neuromodulators, e.g. dopamine, nitric oxide, orexin, neuropeptide Y, and other neuropeptides, are also pivotal in the retina for visual information processing via modulating neuronal activity and synaptic transmission [1,2,3,4,5]. Dysfunction of neuromodulatory systems in the retina may result in retinal diseases, including diabetic retinopathy and glaucoma [6,7,8,9]. Hence, deciphering the expression and function of neuromodulator-related signals in the retina will promote our understanding of the retinal visual system.
Oxytocin is a well-known neurohormone for its function in labor induction and lactation. Oxytocin also acts as a neuropeptide in modulating diverse brain functions, including social, maternal, and emotional behaviors [10,11,12]. Endogenous oxytocin is synthesized and released by oxytocin neurons, which are mainly distributed in the paraventricular nucleus of the hypothalamus (PVN) and supraoptic nucleus (SON). Oxytocin modulates neuronal activity and synaptic transmission via activating the Gq protein-coupled oxytocin receptors (OxtRs), which are broadly expressed in mouse brain areas, including the visual-related brain regions [13, 14]. In the mouse primary visual cortex, oxytocin signaling mediates the experience-dependent cortical development [15] and also modulates spontaneous activity patterns in the develo** visual cortex [16]. Few studies have investigated the involvement of oxytocin signaling in visual information transmission and processing. In the primate visual pathway, OxtRs have been detected in the superior colliculus, pulvinar, and primary visual cortex to modulate gaze direction and attention [17]. Oxytocin has also been detected in the retina of rats, bovines, and humans, and the retinal oxytocin concentration is synchronized with the day/night cycle [18, 19], but the expression and function of oxytocin signal in the retina are still largely unknown. Recent studies observed the OxtR expression in the retinal pigment epithelium (RPE) of humans and rhesus, and they inferred that oxytocin signaling played an important role in the RPE-photoreceptor communication [20, 21]. However, whether OxtRs are functionally expressed in retinal neurons and the role of retinal oxytocin signaling in visual information processing remains largely unknown.
In the mammalian retina, dopamine (DA), released by DA amacrine cells, has multiple neuromodulatory roles in visual functions related to light and contrast adaptation, visual acuity, and circadian rhythmicity [1]. In addition to integrating light inputs from classic photoreceptors and intrinsically photosensitive retinal ganglion cells [22], DA amacrine cells also express some neuromodulator receptors [23] and can be modulated by some neuropeptides, including orexin and neuropeptide Y [5, 24]. Oxytocin signaling regulates the DA system at different levels, including directly modulating DA neuronal activity and DA release via OxtR in the brain [16, 16]. In the visual cortex, OxtR is found to be expressed in the GABAergic interneurons to modulate the excitatory/inhibitory ratio to refine the develo** circuits [16]. As OxtR is also expressed in the GABAergic interneurons in the retina, endogenous oxytocin signal in the retina may act to refine the retinal circuitry during the development. In humans, the role of endogenous oxytocin signals in social perceptual processes, such as perception of facial emotions and visual processing of infant faces [64], has been reported. Enriched social environment, social touch, parental caring, and parent-infant interaction are reported to increase oxytocin levels [15, 61,62,63,64]. Endogenous oxytocin signal in the retina may be involved in promoting social recognition and parental behavior via elevating visual acuity and detection, which requires to be investigated in the future.
In addition to directly regulating neuronal excitability, oxytocin plays a pivotal role in regulating neuronal synaptic transmission via OxtRs expressed in both presynaptic and postsynaptic membranes [13, 16]. In our study, we compared the differences in retinal oxytocin signals between males and females in the following four aspects: (1) Oxtr fluorescence intensities in the INL and GCL are not significantly different between males and females (Additional file 1: Fig. S1A); (2) the numbers of OxtR-eYFP+ neurons from Oxtr-Cre; Ai3 mice in the INL and GCL did not exhibit sex difference (Additional file 1: Fig. S1E); (3) oxytocin-induced retinal DA release was similar in both male and female mice (Additional file 1: Fig. S5D); and (4) intravitreal injection of oxytocin significantly reduced the amplitude of ERG b-wave in both males and females, and the oxytocin-induced ERG b-wave change did not show significant sex difference (Additional file 1: Fig. S6C-S6D). Together, these results indicate that the expression and function of oxytocin signals in adult mouse retinas largely do not exhibit sex differences.
Conclusions
In summary, our study finds that OxtR is functionally expressed in retinal GABAergic ACs, especially in DA subtypes. The possible functions of neuromodulators synthesized by retinal neurons have been extensively investigated [1, 5, 55, 78], our study indicates that neuropeptides synthesized and released from the brain may also involve in regulating retinal visual perception. Though our results suggest activation of OxtR is sufficient to regulate visual information processing, the physiological functions of endogenous oxytocin signal are worth to be investigated in the future. Previous studies found that endogenous oxytocin directly regulates midbrain dopamine neurons to promote pro-social behavior [Tissue processing, immunohistochemistry, and imaging Mice were anesthetized with isoflurane, and the eyes were removed quickly and dissected in 0.1 M phosphate-buffered saline (PBS). Isolated eye cups were fixed in 4% PFA (dissolved in 0.1 M PBS) at 4 °C for 6 h then dehydrated with 10% (w/v) sucrose solution (dissolved in 0.1 M PBS) for 2 h, 20% for 2 h, and 30% for 24 h in sequence. The dehydrated eyes were embedded in OCT and cut at a thickness of 15 μm using a cryostat (CM1950, Leica Microsystems). The sections were mounted onto Superfrost Plus slides (Thermo Fisher Scientific, Waltham, MA) and stored at − 80 °C. For immunostaining to determine neuronal identity, tissues were rinsed with PBS and pretreated in 0.2% Triton-X100 for 1 h at room temperature (RT) then blocked with 0.05% Triton-X100 and 10% bovine serum albumin (BSA) in PBS for 2 h at RT. Tissues were then incubated with primary antibody solution in PBS with 0.2% Triton-X100 and 1% BSA for 2–3 days at 4 °C. After rinsing in PBS three times, the tissues were incubated with secondary antibody solution (goat anti-rabbit 488, 594, 647; goat anti-mouse 647, 1:800; goat anti-guinea pig 647, 1:800; goat anti-sheep 648, 1:800, Life Technologies; donkey anti-goat 594, 1:200, Jackson ImmunoResearch) in PBS for 2 h at RT then dried and covered under glycerol:TBS (3:1) with Hoechst 33,342 (1:1000, Thermo Fisher Scientific). The primary antibodies used in this study include mouse anti-HPC-1 (1:1000, S0664, Sigma), sheep anti-Chx10 (1:1000, ab16141, Abcam), mouse anti-Calbindin (1:1000, CB300, Swant), mouse anti-Brn3a (1:50, MAB1585, Sigma), guinea pig anti-RBPMS (1:500, 43,691, PhosphoSolutions), rabbit anti-Melanopsin (UF008, 1:10,000, AB-N39, ATS), rabbit anti-vasoactive intestinal peptide (VIP, 1:1000, 20,077, Immunostar), rabbit anti-GAD65 + GAD67 (1:1000, ab183999, Abcam), rabbit anti-GABA (1:1000, A2052, Sigma), rabbit anti-GlyT1 (1:1000, AGT-011, Alomone), rabbit anti-PPP1R17 (1:500, HPA047819, the Human Protein Atlas), goat anti-ChAT (1:800, ab144p, Millipore), rabbit anti-tyrosine hydroxylase (TH, 1:1000, ab152, Millipore), mouse anti-TH (1:1000, 22,941, Immunostar), and rabbit anti-CRH (1:100, ab8901, Abcam). The sections were imaged with an Olympus VS120 slide scanning microscope. Confocal images were acquired with a Nikon A1 confocal laser scanning microscope with a × 25 objective. Images were analyzed in ImageJ (FIJI). The retina sections were prepared in the same way as used for immunohistochemistry. Samples were then processed according to the manufacturer’s instructions in the RNAscope Fluorescent Multiplex Assay manual (Advanced Cell Diagnostics, Newark, CA). After finishing smFISH, some samples were further stained with TH or GFP primary antibodies for 24 h at 4 °C then washed and incubated with secondary antibody. Samples were coverslipped with ProLong Gold antifade reagent with DAPI (Molecular Probes). The following probes were used in this study: Oxtr (C1, 406491), Crh (C1, 318931), and EYFP (C3, 312131). Sections were subsequently imaged with a Nikon A1 confocal laser scanning microscope with a × 25 objective lens, with 1 μm between adjacent z-sections. Probe omission or negative probes were carried out as control for every reaction. smFISH images were analyzed as previously reported [25]. Every four adjacent z-stack images were combined. All channels were thresholded to remove background noise. Cellular regions of interest (ROIs) were defined using the GFP IF channel or TH IF channel to localize cell bodies. Since it is not easy to discriminate the single Oxtr punctum within ROIs, the cell in ROI was considered positive for Oxtr when the fluorescence intensity of Oxtr signal within the soma was more than 200 a.u. (based on the negative probe control). All counting experiments were conducted blinded to the experimental group. The mouse retinas were isolated under the microscope, and the retinal pigment epithelium (RPE) was isolated following a previous study [79]. Tissues were lysed with RIPA lysis buffer containing PMSF protease inhibitor (100:1), and total proteins were extracted and protein concentrations were quantified with a bicinchoninic acid (BCA) assay kit (Beyotime Biotech, China). The protein sample’s final concentration was 2 or 3 μg/μl by diluting with sample loading buffer and ddH2O. A total of 20 μg protein was loaded into the polyacrylamide gel and electrophoretically transferred to the polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was blocked for 1 h in 5% non-fat powdered milk and then incubated with primary antibody (rabbit anti-OxtR, 1:2000, AB181077, Abcam) overnight at 4 °C. Mouse anti-GAPDH antibody (1:80,000, 60,004, Proteintech) was used as the control. After incubating with the primary antibody, the PVDF membrane was then rinsed in TBST three times, and incubated with HRP-goat anti-mouse IgG antibody (1:7000, SA00001-1, Proteintech) or HRP-goat anti-rabbit IgG antibody (1:6500, SA00001-2, Proteintech) at room temperature for 1 h. ECL Prime Western Blotting Detection Reagent was used for fluorescence detection by an Odyssey near-infrared imaging scanner (FluorChem E System, Protein Simple, USA). The analysis of images of blots was performed with the AlphaView SA software (Protein Simple, USA). The images of the original uncropped blots have been provided in Additional file 2. The retinas from Oxtr-Cre; Ai3 mice were prepared, and retinal neurons were recorded as previously described [2]. Briefly, mice were dark-adapted for at least 2-h and then anesthetized with 25% urethane (0.2 ml/100 g). The mouse retinas were dissected under dim red light in Ames’ medium (MilliporeSigma) and bubbled with 95% O2 and 5% CO2. The retina was placed in a recording chamber and perfused with oxygenated Ames’ solution at a rate of ~ 3 ml/min. eYFP-labeled neurons in the GCL were visualized using an IR-DIC microscopy. Current-clamp recordings were established with glass pipettes (5–7 MΩ) containing the following (in mM): 120 K-gluconate, 5 NaCl, 4 KCl, 10 HEPES, 2 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 7 Tris-phosphocreatine (pH was adjusted to 7.3); 30 μM D-AP5, 40 μM DNQX, 50 μM L-AP4, 2 μM ACET, 10 μM bicuculline, 10 μM TPMPA, and 10 μM strychnine are used to block NMDA receptors, AMPA receptors, KA receptors, metabotropic glutamate receptors (mGluRs), GABA(A) receptors, GABA(C) receptors, and glycine receptors during recording. 50 μM L-AP4 will completely activate mGluRs to block the further response induced by presynaptic glutamate release in the retina [2, 22, 31]. Both spontaneous activity and current injection-induced responses were recorded before and during the application of 1 μM oxytocin. Data were obtained using an Axon 700B amplifier, digitized at 10 kHz, filtered at 4 kHz, and collected using the pCLAMP software (Molecular Devices). Animals were dark-adapted for 24 h and then deeply anesthetized with 0.6% pentobarbital sodium (15 μl/g). One microliter oxytocin (1 mM, 500 μM, 100 μM, 10 μM, and 1 μM) or 1 μl WAY267464 (1 mM), dissolved with saline solution, was injected into the vitreous of one eye by a NanojectIII microinjector (Drummond Scientific Company, USA) at a speed of ~ 5 nl/s, and the other eye was injected with the same volume of saline as control. The 1 μl volume was chosen based on a previous study [2]. Since 1 mM oxytocin application had a reliable and large effect on the reduction of ERG b-wave, 1 mM oxytocin was used for the experiments to investigate the possible mechanisms. To further verify the results of exogenous oxytocin application, the specific and selective OxtR agonist WAY267464 [25, 80] was used, which has been widely used as the non-peptide agonist to investigate the function of oxytocin signal. About 1 h after oxytocin and saline injection, the retinas or vitreous bodies (collected by an Eppendorf pipette with a 10 μl pipette tip) were harvested [42]. As described previously [81], each frozen sample was homogenized into 100 μl of ice-cold 0.1 M perchloric acid containing 10 μM ascorbic acid, 0.1 mM EDTA disodium salt, and 0.02 μM 3,4-dihydroxybenzyl-amine. Dopamine, DOPAC (3,4-dihydroxyphenylacetic acid), and HVA (Homovanillic acid) levels were measured with the Agilent 1200 series neurotransmitter analyzer (Agilent Technologies, Santa Clara, CA, USA). Data were collected and analyzed by ChemStation (Agilent Technologies). For the cFos immunostaining experiment, mice were exposed to the environment with light intensity at ~ 1.0 cd/m2 for 1 h, and mice were anesthetized during the whole procedure. Then, mice were sacrificed and the retinas were dissected, fixed, and sliced for cFos immunostaining as described previously. Rabbit anti-cFos (1:1000, 5348, Cell Signaling) and mouse anti-TH (1:1000, 22,941, Immunostar) were used to detect cFos expression in retinal DA neurons. To assess retinal function, ~ 30 min after the intravitreal injection, ERG was measured as previously reported [81]. The whole procedure was conducted in darkness. Mice were kept anesthetized with 0.6% pentobarbital sodium, and the pupils were dilated by compound tropicamide eye drops (Mydrin-p, Santen Pharmaceutical, Japan). Gold wire ring electrodes (3104RC, Roland, Germany) were placed onto the surface of both corneas, and ERGs were acquired by a pre-amplifier (FZG-81, Jia Long Educational Instruments, China) and band-pass filtered (0.1–100 Hz); 3-ms white light flashes were generated by a LED light source (CQ-LU9079, Qianhan Lighting, China) and presented by a custom-built Ganzfeld dome with 5 different stimulus strengths (0.0016 cd·s/m2, 0.0388 cd·s/m2, 0.31 cd·s/m2, 1.47 cd·s/m2, and 2.65 cd·s/m2). Light stimulation was controlled by a multi-data acquisition card (PCIe 6321, National Instruments, USA) with a LabVIEW-based code. The animals were placed on a thermostatic plate to maintain body temperature during the recording. The amplitudes of ERG a-wave and b-wave were analyzed after recording. OxtR antagonist—L368, 899 (5 mg/kg); vasopressin 1a receptor antagonist—SR49059 (10 mg/kg); dopamine D1 receptor antagonist—SCH23390 (5 mg/kg); or dopamine D2 receptor antagonist—L741, 626 (3 mg/kg)—were given intraperitoneally ~ 30 min prior to the intravitreal injection of oxytocin. The dosage for L368, 899; SR49059; SCH23390; and L741, 626 was determined from previous studies in mice and rats [82,83,84,85]. All image analyses were carried out in ImageJ (FIJI, NIH). The number of neurons and the number of animals used in every experiment are provided in the figure legends. Group data are expressed as the mean ± SEM. Statistical analysis was performed in GraphPad Prism (GraphPad). Normality was evaluated by the Kolmogorov–Smirnov normality test using GraphPad Prism. For two-group comparisons, statistical significance was determined by two-tailed paired or unpaired Student’s t-tests, and Wilcoxon signed-rank test or Mann–Whitney test when assumptions for parametric testing were not satisfied. For multiple group comparisons, two-way and one-way analyses of variance (ANOVA) tests were used for normally distributed data, followed by post hoc analyses. For data that were not normally distributed, non-parametric tests for the appropriate group types were used instead. p < 0.05 was considered statistically significant.Quantitative fluorescence single-molecule in situ hybridization (smFISH)
Western blot analysis
Electrophysiological recording
Intravitreal injection
Measuring retinal dopamine level with high-performance liquid chromatography (HPLC)
cFos immunostaining and electroretinographic (ERG) recording
Quantification and statistical analysis
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- a.u.:
-
Arbitrary units
- AC:
-
Amacrine cell
- ChAT:
-
Cholinergic
- CRH:
-
Corticotrophin-releasing hormone
- DA:
-
Dopamine
- DOPAC:
-
3,4-Dihydroxyphenylacetic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- ERG:
-
Electroretinographic
- eYFP:
-
Enhanced yellow fluorescent protein
- FISH:
-
Fluorescent in situ hybridization
- GABA:
-
Gamma-aminobutyric acid
- GCL:
-
Ganglion cell layer
- GFP:
-
Green fluorescent protein
- HC:
-
Horizontal cell
- HVA:
-
Homovanillic acid
- i.p.:
-
Intraperitoneal
- IF:
-
Immunofluorescence
- INL:
-
Inner nuclear layer
- IPL:
-
Inner plexiform layer
- ns:
-
Not significant
- ONL:
-
Outer nuclear layer
- OPL:
-
Outer plexiform layer
- Oxt:
-
Oxytocin
- OxtR:
-
Oxytocin receptor
- PBS:
-
Phosphate-buffered saline
- PFA:
-
Paraformaldehyde
- PVN:
-
Paraventricular nucleus of the hypothalamus
- RGC:
-
Retinal ganglion cell
- RPE:
-
Retinal pigment epithelium
- SON:
-
Supraoptic nucleus
- WB:
-
Western blot
- SAL:
-
Saline
- TBS:
-
Tris-buffered saline
- TH:
-
Tyrosine hydroxylase
- VIP:
-
Vasoactive intestinal peptide
References
Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40.
Zhou W, Wang LQ, Shao YQ, Han X, Yu CX, Yuan F, et al. Orexin-a intensifies mouse pupillary light response by modulating intrinsically photosensitive retinal ganglion cells. J Neurosci. 2021;41:2566–80.
Martins J, Elvas F, Brudzewsky D, Martins T, Kolomiets B, Tralhão P, et al. Activation of neuropeptide Y receptors modulates retinal ganglion cell physiology and exerts neuroprotective actions in vitro. ASN Neuro. 2015;7:1759091415598292.
Jacoby J, Nath A, Jessen ZF, Schwartz GW. A self-regulating gap junction network of amacrine cells controls nitric oxide release in the retina. Neuron. 2018;100:1149-1162.e5.
Santos-Carvalho A, Álvaro AR, Martins J, Ambrósio AF, Cavadas C. Emerging novel roles of neuropeptide Y in the retina: from neuromodulation to neuroprotection. Prog Neurobiol. 2014;112:70–9.
Kim MK, Aung MH, Mees L, Olson DE, Pozdeyev N, Iuvone PM, et al. Dopamine deficiency mediates early rod-driven inner retinal dysfunction in diabetic mice. Investig Ophthalmol Vis Sci. 2018;59:572–81.
Motz CT, Chesler KC, Allen RS, Bales KL, Mees LM, Feola AJ, et al. Novel detection and restorative levodopa treatment for preclinical diabetic retinopathy. Diabetes. 2020;69:1518–27.
Polak K, Luksch A, Berisha F, Fuchsjaeger-Mayrl G, Dallinger S, Schmetterer L. Altered nitric oxide system in patients with open-angle glaucoma. Arch Ophthalmol. 2007;125:494–8.
Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci U S A. 1999;96:9944–8.
Neumann ID, Landgraf R. Tracking oxytocin functions in the rodent brain during the last 30 years: from push-pull perfusion to chemogenetic silencing. J Neuroendocrinol. 2019;31:e12695.
Lee HJ, Macbeth AH, Pagani JH, Scott YW. Oxytocin: the great facilitator of life. Prog Neurobiol. 2009;88:127–51.
Froemke RC, Young LJ. Oxytocin, neural plasticity, and social behavior. Annu Rev Neurosci. 2021;44:359–81.
Mitre M, Marlin BJ, Schiavo JK, Morina E, Norden SE, Hackett TA, et al. A distributed network for social cognition enriched for oxytocin receptors. J Neurosci. 2016;36:2517–35.
Grinevich V, Stoop R. Interplay between oxytocin and sensory systems in the orchestration of socio-emotional behaviors. Neuron. 2018;99:887–904.
Zheng JJ, Li SJ, Zhang XD, Miao WY, Zhang D, Yao H, et al. Oxytocin mediates early experience-dependent cross-modal plasticity in the sensory cortices. Nat Neurosci. 2014;17:391–9.
Maldonado PP, Nuno-Perez A, Kirchner JH, Hammock E, Gjorgjieva J, Lohmann C. Oxytocin shapes spontaneous activity patterns in the develo** visual cortex by activating somatostatin interneurons. Curr Biol. 2021;31:322-333.e5.
Freeman SM, Young LJ. Comparative perspectives on oxytocin and vasopressin receptor research in rodents and primates: translational implications. J Neuroendocrinol. 2016;28:10.
Gauquelin G, Geelen G, Louis F, Allevard AM, Meunier C, Cuisinaud G, et al. Presence of vasopressin, oxytocin and neurophysin in the retina of mammals, effect of light and darkness, comparison with the neuropeptide content of the neurohypophysis and the pineal gland. Peptides. 1983;4:509–15.
Gauquelin G, Gharib C, Ghaemmaghami F, Allevard AM, Cherbal F, Geelen G, et al. A day/night rhythm of vasopressin and oxytocin in rat retina, pineal and harderian gland. Peptides. 1988;9:289–93.
Halbach P, Pillers DAM, York N, Asuma MP, Chiu MA, Luo W, et al. Oxytocin expression and function in the posterior retina: a novel signaling pathway. Investig Ophthalmol Vis Sci. 2015;56:751–60.
York N, Halbach P, Chiu MA, Bird IM, Pillers DAM, Pattnaik BR. Oxytocin (OXT)-stimulated inhibition of Kir7.1 activity is through PIP2-dependent Ca2 + response of the oxytocin receptor in the retinal pigment epithelium in vitro. Cell Signal. 2017;37:93–102.
Zhang DQ, Wong KY, Sollars PJ, Berson DM, Pickard GE, McMahon DG. Intraretinal signaling by ganglion cell photoreceptors to dopaminergic amacrine neurons. Proc Natl Acad Sci U S A. 2008;105:14181–6.
Yan W, Laboulaye MA, Tran NM, Whitney IE, Benhar I, Sanes JR. Mouse retinal cell atlas: molecular identification of over sixty amacrine cell types. J Neurosci. 2020;40:5177–95.
Qiao SN, Zhou W, Liu LL, Zhang DQ, Zhong YM. Orexin-a suppresses signal transmission to dopaminergic amacrine cells from outer and inner retinal photoreceptors. Investig Ophthalmol Vis Sci. 2017;58:4712–21.
**ao L, Priest MF, Nasenbeny J, Lu T, Kozorovitskiy Y. Biased oxytocinergic modulation of midbrain dopamine systems. Neuron. 2017;95:368-384.e5.
**ao L, Priest MF, Kozorovitskiy Y. Oxytocin functions as a spatiotemporal filter for excitatory synaptic inputs to VTA dopamine neurons. Elife. 2018;7:e33892.
Hung LW, Neuner S, Polepalli JS, Beier KT, Wright M, Walsh JJ, et al. Gating of social reward by oxytocin in the ventral tegmental area. Science (80- ). 2017;357:1406–11.
Love TM. Oxytocin, motivation and the role of dopamine. Pharmacol Biochem Behav. 2014;119:49–60.
Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–83.
Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, et al. A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell. 2018;174:465-480.e22.
Sonoda T, Lee SK, Birnbaumer L, Schmidt TM. Melanopsin phototransduction is repurposed by ipRGC subtypes to shape the function of distinct visual circuits. Neuron. 2018;99:754-767.e4.
Masland RH. The neuronal organization of the retina. Neuron. 2012;76:266–80.
Schmidt TM, Chen SK, Hattar S. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 2011;34:572–80.
Wassle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–57.
Masland RH. The tasks of amacrine cells. Vis Neurosci. 2012;29:3–9.
MacNeil M, Masland RH. Extreme diversity among amacrine cells: implications for function. Neuron. 1998;20:971–82.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14.
Cui LJ, Chen WH, Liu AL, Han X, Jiang SX, Yuan F, et al. NGnG amacrine cells and Brn3b-negative M1 ipRGCs are specifically labeled in the ChAT-ChR2-EYFP mouse. Investig Ophthalmol Vis Sci. 2020;61:14.
Park SJH, Pottackal J, Ke JB, Jun NY, Rahmani P, Kim IJ, et al. Convergence and divergence of CRH amacrine cells in mouse retinal circuitry. J Neurosci. 2018;38:3753–66.
Pérez de Sevilla Müller L, Solomon A, Sheets K, Hapukino H, Rodriguez AR, Brecha NC. Multiple cell types form the VIP amacrine cell population. J Comp Neurol. 2019;527:133–58.
Li S, Sukeena JM, Simmons AB, Hansen EJ, Nuhn RE, Samuels IS, et al. DSCAM promotes refinement in the mouse retina through cell death and restriction of exploring dendrites. J Neurosci. 2015;35:5640–54.
Liu AL, Liu YF, Wang G, Shao YQ, Yu CX, Yang Z, et al. The role of ipRGCs in ocular growth and myopia development. Sci Adv. 2022;8:eabm9027.
Megaw P, Morgan I, Boelen M. Vitreal dihydroxyphenylacetic acid (DOPAC) as an index of retinal dopamine release. J Neurochem. 2001;76:1636–44.
Smith BJ, Côté PD, Tremblay F. Dopamine modulation of rod pathway signaling by suppression of GABAC feedback to rod-driven depolarizing bipolar cells. Eur J Neurosci. 2015;42:2258–70.
Travis AM, Heflin SJ, Hirano AA, Brecha NC, Arshavsky VY. Dopamine-dependent sensitization of rod bipolar cells by GABA is conveyed through wide-field amacrine cells. J Neurosci. 2018;38:723–32.
Stockton RA, Slaughter MM. B-wave of the electroretinogram: a reflection of ON bipolar cell activity. J Gen Physiol. 1989;93:101–22.
Miura G, Wang MH, Ivers KM, Frishman LJ. Retinal pathway origins of the pattern ERG of the mouse. Exp Eye Res. 2009;89:49–62.
Kaplan HJ, Chiang CW, Chen J, Song SK. Vitreous volume of the mouse measured by quantitative high-resolution MRI. Invest Ophthalmol Vis Sci. 2010;51:4414.
Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, et al. Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol. 2012;24:609–28.
Tian N, Xu HP, Wang P. Dopamine D2 receptors preferentially regulate the development of light responses of the inner retina. Eur J Neurosci. 2015;41:17–30.
Gottvall E, Textorius O. Concentration-dependent effects of dopamine on the direct current electroretinogram of pigmented rabbits during prolonged intermittent recording. Doc Ophthalmol. 2003;106:161–9.
Chaffiol A, Ishii M, Cao Y, Mangel SC. Dopamine regulation of GABAA receptors contributes to light/dark modulation of the ON-cone bipolar cell receptive field surround in the retina. Curr Biol. 2017;27:2600-2609.e4.
Hellmer CB, Bohl JM, Hall LM, Koehler CC, Ichinose T. Dopaminergic modulation of signal processing in a subset of retinal bipolar cells. Front Cell Neurosci. 2020;14:1–14.
Nguyen-Legros J, Versaux-Botteri C, Vernier P. Dopamine receptor localization in the mammalian retina. Mol Neurobiol. 1999;19:181–204.
Roy S, Field GD. Dopaminergic modulation of retinal processing from starlight to sunlight. J Pharmacol Sci. 2019;140:86–93.
Zhou ZJ, Fain GL. Starburst amacrine cells change from spiking to nonspiking neurons during retinal development. Proc Natl Acad Sci U S A. 1996;93:8057–62.
Csáki Á, Köves K, Boldogkői Z, Tombácz D, Tóth ZE. The same magnocellular neurons send axon collaterals to the posterior pituitary and retina or to the posterior pituitary and autonomic preganglionic centers of the eye in rats. NeuroSci. 2021;2:27–44.
Yamamoto Y, Liang M, Munesue S, Deguchi K, Harashima A, Furuhara K, et al. Vascular RAGE transports oxytocin into the brain to elicit its maternal bonding behaviour in mice. Commun Biol. 2019;2:76.
Munesue S, Liang MK, Harashima A, Zhong J, Furuhara K, Boitsova EB, et al. Transport of oxytocin to the brain after peripheral administration by membrane-bound or soluble forms of receptors for advanced glycation end-products. J Neuroendocrinol. 2021;33:e12963.
McVicar CM, Ward M, Colhoun LM, Guduric-Fuchs J, Bierhaus A, Fleming T, et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia. 2015;58:1129–37.
Tang Y, Benusiglio D, Lefevre A, Hilfiger L, Althammer F, Bludau A, et al. Social touch promotes interfemale communication via activation of parvocellular oxytocin neurons. Nat Neurosci. 2020;23:1125–37.
Yu H, Miao W, Ji E, Huang S, ** S, Zhu X, et al. Social touch-like tactile stimulation activates a tachykinin 1-oxytocin pathway to promote social interactions. Neuron. 2022;110:1051-1067.e7.
Carcea I, Caraballo NL, Marlin BJ, Ooyama R, Riceberg JS, Mendoza Navarro JM, et al. Oxytocin neurons enable social transmission of maternal behaviour. Nature. 2021;596:553–7.
Feldman R, Gordon I, Schneiderman I, Weisman O, Zagoory-Sharon O. Natural variations in maternal and paternal care are associated with systematic changes in oxytocin following parent-infant contact. Psychoneuroendocrinology. 2010;35:1133–41.
Dief AE, Sivukhina EV, Jirikowski GF. Oxytocin and stress response. Open J Endocr Metab Dis. 2018;08:93–104.
Bakos J, Srancikova A, Havranek T, Bacova Z. Molecular mechanisms of oxytocin signaling at the synaptic connection. Neural Plast. 2018;2018:4864107.
Yoshida K, Watanabe D, Ishikane H, Tachibana M, Pastan I, Nakanishi S. A key role of starburst amacrine cells in originating retinal directional selectivity and optokinetic eye movement. Neuron. 2001;30:771–80.
Li Y, Turan Z, Meister M. Functional architecture of motion direction in the mouse superior colliculus. Curr Biol. 2020;30:3304-3315.e4.
Shi X, Barchini J, Ledesma HA, Koren D, ** Y, Liu X, et al. Retinal origin of direction selectivity in the superior colliculus. Nat Neurosci. 2017;20:550–8.
Basso MA, Bickford ME, Cang J. Unraveling circuits of visual perception and cognition through the superior colliculus. Neuron. 2021;109:918–37.
Tran VT, Dickman M. Differential localization of dopamine D1 and D2 receptors in rat retina. Investig Ophthalmol Vis Sci. 1992;33:1620–6.
Jagadeesh JM, Sanchez R. Effects of apomorphine on the rabbit electroretinogram. Invest Ophthalmol Vis Sci. 1981;21:620–5.
Li K, Nakajima M, Ibañez-Tallon I, Heintz N. A cortical circuit for sexually dimorphic oxytocin-dependent anxiety behaviors. Cell. 2016;167:60-72.e11.
Francis DD, Young LJ, Meaney MJ, Insel TR. Naturally occurring differences in maternal care are associated with the expression of oxytocin and vasopressin (V1a) receptors: gender differences. J Neuroendocrinol. 2002;14:349–53.
Borland JM, Aiani LM, Norvelle A, Grantham KN, O’Laughlin K, Terranova JI, et al. Sex-dependent regulation of social reward by oxytocin receptors in the ventral tegmental area. Neuropsychopharmacology. 2019;44:785–92.
Chen S, Xu H, Dong S, **ao L. Morpho-electric properties and diversity of oxytocin neurons in paraventricular nucleus of hypothalamus in female and male mice. J Neurosci. 2022;42:2885–904.
Son S, Manjila SB, Newmaster KT, Wu Y, Vanselow DJ, Ciarletta M, et al. Whole-brain wiring diagram of oxytocin system in adult mice. J Neurosci. 2022;42:5021–33.
Vielma AH, Retamal MA, Schmachtenberg O. Nitric oxide signaling in the retina: what have we learned in two decades? Brain Res. 2012;1430:112–25.
Wei H, Xun Z, Granado H, Wu A, Handa JT. An easy, rapid method to isolate RPE cell protein from the mouse eye. Exp Eye Res. 2016;145:450–5.
Hicks C, Jorgensen W, Brown C, Fardell J, Koehbach J, Gruber CW, et al. The nonpeptide oxytocin receptor agonist WAY 267,464: receptor-binding profile, prosocial effects and distribution of c-Fos expression in adolescent rats. J Neuroendocrinol. 2012;24:1012–29.
Wu XH, Qian KW, Xu GZ, Li YY, Ma YY, Huang F, et al. The role of retinal dopamine in C57BL/6 mouse refractive development as revealed by intravitreal administration of 6-hydroxydopamine. Investig Ophthalmol Vis Sci. 2016;57:5393–404.
Choe HK, Reed MD, Benavidez N, Montgomery D, Soares N, Yim YS, et al. Oxytocin mediates entrainment of sensory stimuli to social cues of opposing valence. Neuron. 2015;87:152–63.
Tan O, Musullulu H, Raymond JS, Wilson B, Langguth M, Bowen MT. Oxytocin and vasopressin inhibit hyper-aggressive behaviour in socially isolated mice. Neuropharmacology. 2019;156:107573.
Mohd-Yusof A, Gonzalez AE, Veliz A, McDougall SA. Role of the D1 receptor for the dopamine agonist-induced one-trial behavioral sensitization of preweanling rats. Psychopharmacology. 2014;231:4167–77.
Zhang HY, Shen H, Gao M, Ma Z, Hempel BJ, Bi GH, et al. Cannabinoid CB2 receptors are expressed in glutamate neurons in the red nucleus and functionally modulate motor behavior in mice. Neuropharmacology. 2021;189:108538.
Acknowledgements
We thank Dr. Lan Ma for the CRH antibody and Crh probe, Dr. Yongchun Yu for the GABA antibody, Dr. Yilin Tai for the Calbindin antibody, Dr. Miao He for the Ai3 mice, and Dr. Peiji Liang and the members of **ao laboratory for their valuable input. This work was supported by grants from the National Natural Science Foundation of China (81970727, 31900738), the Lingang Laboratory (LG-QS-202203-12), the Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01), the ZJ Lab, and the Shanghai Center for Brain Science and Brain-Inspired Technology.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81970727, 31900738), the Lingang Laboratory (LG-QS-202203–12), the Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01), the ZJ Lab, and the Shanghai Center for Brain Science and Brain-Inspired Technology.
Author information
Authors and Affiliations
Contributions
LX designed and supervised the project. SH and YW did the experiments and analyzed the data. XH and SW conducted the patch-clamp recording. MD and YZ contributed to the electrophysiological recording and image analyses. YM contributed to the HPLC experiments. The manuscript was written by LX, SH, and YW. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animals were handled following the protocols approved by the Fudan University Animal Care and Use Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The expression of OxtRs in mouse retina. Fig. S2. Example images showing OxtR-eYFP neurons are Brn3a (A) or melanopsin (B) positive in the GCL. Fig. S3. eYFP+ neurons in Oxtr-Cre; Ai3 mice are mainly GABAergic, but not glycinergic amacrine cells. Fig. S4. Distribution of OxtR-eYFP neurons in Oxtr-Cre; Ai3 mouse retina. Fig. S5. ChAT and CRH ACs are partially co-localized with OxtR-eYFP neurons, and oxytocin elevates DA level. Fig. S6. The effects of oxytocin and OxtR antagonist on the amplitude of ERG b-wave. Fig. S7. Effects of oxytocin on b-wave amplitude in the presence of V1AR or DA receptor antagonists.
Additional file 2.
The images of the original, uncropped blots for the OxtR and GAPDH.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, S., Wang, Y., Han, X. et al. Activation of oxytocin receptors in mouse GABAergic amacrine cells modulates retinal dopaminergic signaling. BMC Biol 20, 205 (2022). https://doi.org/10.1186/s12915-022-01405-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-022-01405-0