
Abstract
Background
Individuals born with cleft lip and/or palate who receive corrective surgery regularly have abnormal growth in the midface region such that they exhibit premaxillary hypoplasia. However, there are also genetic contributions to craniofacial morphology in the midface region, so although these individuals appear to have Class III skeletal discrepancy, their molar relationship may be Class I. Past genome-wide association studies (GWASs) on skeletal Class II and III malocclusion suggested that multiple genetic markers contribute to these phenotypes via a multifactorial inheritance model, but research has yet to examine the genetic markers associated with dental Class I malocclusion. Thus, our goal was to conduct a family based GWAS to identify genes across the genome that are associated with Class I malocclusion, as defined by molar relations, in humans with and without clefts.
Methods
Our cohort consisted of 739 individuals from 47 Filipino families originally recruited in 2006 to investigate the genetic basis of orofacial clefts. All individuals supplied blood samples for DNA extraction and genoty**, and a 5,766 single nucleotide polymorphism (SNP) custom panel was used for the analyses. We performed a transmission disequilibrium test for participants with and without clefts to identify genetic contributors potentially involved with Class I malocclusion.
Results
In the total cohort, 13 SNPs had associations that reached the genomic control threshold (p < 0.005), while five SNPs were associated with Class I in the cohort of participants without clefts, including four associations that were identified in the total cohort. The associations for the SNPs ABCA4 rs952499, SOX1-OT rs726455, and RORA rs877228 are of particular interest, as past research found associations between these genes and various craniofacial phenotypes, including cleft lip and/or palate.
Conclusions
These findings support the multifactorial inheritance model for dental Class I malocclusion and suggest a common genetic basis for different aspects of craniofacial development.
Similar content being viewed by others


Introduction
The “father of modern orthodontics”, Dr. Edward Hartley Angle determined three types of malocclusion: Class I, II, and III [1]. In the present study, we are interested in the Class I molar classification or “neutroclusion”, which is established as the mesiobuccal cusp of the maxillary first molar occluding with the buccal grove of the mandibular first molar [1, 2]. It has been suggested that 92% of malocclusion cases have unknown etiology and genetics may play a significant role in craniofacial morphology of the midface region [2,3,4].
Cleft lip and/or palate is a common condition that affects about 1 in every 600 newborn babies. It consists of a failure in the closure of fetal facial tissues, including lip and/or palate tissues, prenatally [5]. Individuals born with cleft lip and/or palate who receive corrective surgery regularly have disrupted growth in the midface region such that they may exhibit premaxillary hypoplasia, crossbites, and/or malocclusion [6, 7].
Past genome-wide association studies (GWASs) on skeletal Class II and III malocclusion suggested that multiple genetic markers contribute to these phenotypes via a multifactorial inheritance model [4], but research has yet to examine the genetic markers associated with dental Class I malocclusion in individuals with or without clefts.
In the present study, we conducted a family-based GWAS in individuals affected by dental Class I malocclusion, as defined by molar relations, and clefts. We aimed to identify genes across the genome that are associated with Class I malocclusion in 47 Filipino families in order to gain insight into the genetic basis of this favorable craniofacial phenotype in humans with and without clefts.
Methods
Participants
Our study cohort consisted of 739 individuals (377 males and 362 females) from 47 families who were small-scale fishermen or landless rural dwellers residing in the central region of the Philippines, mainly Cebu Island, and the surrounding islands. This cohort was previously obtained for the purpose of investigating orofacial characteristics and genetic profiles of individuals who had similar cultural backgrounds and who were exposed to similar environmental factors [8]. Since the individuals in the cohort had shared variables of similar cultural backgrounds and the same area of residence, their access to care and their environmental influences were also similar, reducing possible confounders.
The original phenotype examined in the cohort was orofacial clefts. Informed consent was obtained from all participants and the project had appropriate approvals from Institutional Review Boards. All participants supplied blood samples for DNA extraction and genoty** and underwent a clinical evaluation by an experienced dentist to assess whether they had Class I malocclusion. The clinical criterion for making the diagnosis was a Class I molar relationship based on a visual clinical examination of the individual’s molar-canine relationships where the mesiobuccal cusp of the maxillary first molar should be occluding with the buccal grove of the mandibular first molar [2].
One hundred and nine individuals in the cohort were diagnosed with Class I malocclusion (55 males and 54 females). In the present study, 90 of the 739 individuals included in the analysis had clefts and only six individuals were diagnosed with Class I malocclusion and clefts concurrently. There was not a large diversity of ages within the sample population. Most of the participants were young, in their twenties, after facial growth is complete. Further oral conditions that were identified in this population and are common in the general population include gingivitis, plaque, dental caries, and torus palatinus.
Genomic analyses
A custom panel including 5,766 single nucleotide polymorphisms (SNPs) was previously used to generate genoty** data at the Center for Inherited Disease Research (CIDR). The criteria for selecting the custom panel included the minor allele frequency of the SNP, the location of the SNP in the gene, the postulated function of the gene, and the linkage disequilibrium structure of a genomic region that accounts for proximate genes. The PedCheck program was utilized to test for inconsistencies in the data due to non-paternity or other errors [9].
In the present study, we performed a family-based transmission disequilibrium test (TDT) using PLINK software [10], which tested for the over-transmission of alleles of the 5,766 markers and Class I malocclusion in the cohort. Since the TDT measures the transmission of marker alleles of heterozygous parents to the affected offspring and the non-transmitted alleles act as controls to the transmitted ones [11], the test is suitable and can avoid the issue of potential deceptive associations that can be present in case-control studies. We conducted data analysis in the total sample and in a subset of the sample that excluded individuals with clefts, as both Class I malocclusion and clefts affect the same or adjacent craniofacial structures, and the additional analysis could distinguish genetic contributors that also contribute to clefts. In addition, we generated adjusted significance values to account for multiple testing. Since the Bonferroni correction and the false discovery rate (FDR) correction were too stringent, we also report the genomic control (GC) correction to set our significance threshold of p < 0.005. Given our limited sample of individuals with Class I (109 people) and because we did not want to risk not identifying relevant biological associations for this understudied phenotype, we report GC (a more relaxed correction for multiple testing, p < 0.005). In the present study, the Bonferroni correction sets the significance threshold to p-value < 8.7 × 10− 6.
Additional analyses for sex differences and Class I phenotype presence (affected and unaffected) according to each genotype were performed for the most relevant results found in the aforementioned TDT analysis (GC p < 0.005).
Results
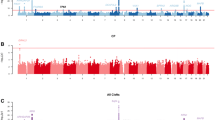
Following the completion of the TDT for the 47 families in the total cohort, we discovered that there were relevant SNPs (GC corrected p value < 0.005) present in chromosomes 1, 3, 4, 5, 10, 11, 12, 13, 14, 15, 20, and 23. The SNPs with the lowest GC corrected p values associated with increased likelihood of Class I malocclusion development were as follows: rs952499 (ABCA4), rs3847993 (IL17D), and rs726455 (SOX1-OT) (Table 1). When we completed the TDT tests in the subset of the sample excluding individuals with clefts, only four of the associations remained under the GC p < 0.005 significance level. However, a new association was identified in chromosome 1 for the SNP rs1060622 (TMED5) (Table 2). None of the tested SNPs reached the genome-wide significance threshold when either the FDR or the Bonferroni correction were applied.
The prevalence of Class I malocclusion in our total cohort is 14.7%, where 50.5% of affected individuals are males and 49.5% are females. For the subset of individuals in the total cohort who have Class I malocclusion and not clefts, the prevalence of Class I malocclusion is 15.9%, where 49.5% are males and 50.5% are females. We broke down the GC significant genoty** results by sex in the total cohort and in the cohort of participants with Class I malocclusion without clefts and report the most relevant (GC p < 0.005) results in Table 3. The rs1874925 in SYNPR reached GC significance threshold in the total cohort with females more frequently carrying the homozygous for the major allele (CC) (Table 3). When breaking down the genoty** results by Class I status and comparing the cases and the comparison group, no significant associations were identified. Using the presence of oral conditions including dental caries as a covariate also did not affect the results identified.
Discussion
In the present study, we report various associations between SNPs within the genome and Class I malocclusion in participants with and without clefts which have not yet been examined in the literature. Two significant associations found in this study were for SNPs in the genes ABCA4, which encodes the membrane-associated protein ATP-binding cassette transporter [12], and SOX1-OT, which is a non-coding RNA that regulates gene expression [13]. Previous research showed that ABCA4 is associated with craniofacial skeletal variation among patients with skeletal malocclusion [14], and many studies have found associations between ABCA4 and SOX1-OT and cleft lip with or without cleft palate [15,16,17,18,19]. The associations of these genes with skeletal malocclusion and clefts in previous research and with dental malocclusion in our project, despite the fact that we are focusing on dental malocclusion rather than skeletal malocclusion, indicates that there may be a shared genetic basis for various aspects of craniofacial growth and development.
Another significant association identified in this study was for a SNP in the gene RORA, which encodes for the nuclear hormone receptor RAR related orphan receptor A [20]. This gene has been shown to be associated with a risk of orofacial clefts [21] and neuropathy in individuals with head and neck cancer [22]. RORA was also found to exhibit significantly decreased transcript expression in dental pulp stem cells from subjects with 15q Duplication syndrome compared to controls [23]. Individuals with this syndrome can have unusual facial features such as micrognathia, a high-arched palate, a long philtrum, and anteverted nares [24]. The IL17D association was a novel finding and there is a lack of literature describing implications related to this gene.
Additionally, we broke down the significant genoty** results by sex in the affected patients from the total cohort and identified significant differences in the frequency of the rs1874925 in SYNPR between males and females.
We are aware that the small sample size and the specificity of the Filipino cohort limit the generalizability of our study. We therefore need to confirm our findings using a larger sample and multiple populations that are exposed to various environmental influences. However, the cohort’s shared exposure to the same environmental factors during data collection reduced the heterogeneity of the sample, boosted confidence that the control group accurately represented the affected group, and increased the likelihood of discovering biologically relevant results. It is also relevant to note that three methods for correction were implemented: GC, FDR, and Bonferroni. Statistically significant results were found only when the genomic control correction [25] was used. This is likely due to the fact that GC controls for confounding effects of population stratification, based on the distribution of the chi-square statistics for the allele suspected to be associated with the phenotype compared to the same distribution of another allele not associated with the phenotype in a different chromosome. It is important to highlight that GC may be a too relaxed multiple testing approach and our results should be taken with caution since that false positives are a possibility.
Our study was not only limited by the small sample size but also the unremarkable age distribution of the participants (patients and family members were often young). It is necessary to corroborate our findings with the addition of age as a variable. Additionally, in future studies, it would be interesting to investigate the association between Class I malocclusion with the different subtypes of clefts. Since in our study only six people had both clefts and Class I malocclusion diagnoses, we could not accurately investigate this relationship.
In conclusion, this study adds to the body of knowledge on genetics of malocclusion and cleft lip and/or palate by showing that several SNPs previously linked to a variety of craniofacial phenotypes are linked to a Class I molar relationship. The notion of a shared genetic basis for disparate elements of craniofacial development is intriguing and further research should explore this possibility as well as the processes by which these genes may interact to cause the observed phenotypes.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Proffit WR, Fields HW, Sarver DM. Contemporary orthodontics. 4th ed. St. Louis, Mo.: Mosby Elsevier; 2007. xii, 751 p. p.
Ghodasra R, Brizuela M, Orthodontics. Malocclusion. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Melina Brizuela declares no relevant financial relationships with ineligible companies.2023.
Rolfe S, Lee SI, Shapiro L. Associations between genetic data and quantitative Assessment of normal facial asymmetry. Front Genet. 2018;9:659.
Gershater E, Li C, Ha P, Chung CH, Tanna N, Zou M et al. Genes and pathways associated with skeletal sagittal malocclusions: a systematic review. Int J Mol Sci. 2021;22(23).
Vyas T, Gupta P, Kumar S, Gupta R, Gupta T, Singh HP. Cleft of lip and palate: a review. J Family Med Prim Care. 2020;9(6):2621–5.
Paradowska-Stolarz A, Kawala B. Occlusal disorders among patients with total clefts of lip, alveolar bone, and palate. Biomed Res Int. 2014;2014:583416.
Alkhouri S, Waite PD, Davis MB, Lamani E, Kau CH. Maxillary distraction osteogenesis in unilateral cleft lip and palate patients with rigid external distraction system. Ann Maxillofac Surg. 2017;7(1):57–63.
Vieira AR, Marazita ML, Goldstein-McHenry T. Genome-wide scan finds suggestive caries loci. J Dent Res. 2008;87(5):435–9.
O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63(1):259–66.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Floros J, Fan R, Matthews A, DiAngelo S, Luo J, Nielsen H, et al. Family-based transmission disequilibrium test (TDT) and case-control association studies reveal surfactant protein A (SP-A) susceptibility alleles for respiratory distress syndrome (RDS) and possible race differences. Clin Genet. 2001;60(3):178–87.
Whelan L, Dockery A, Stephenson KAJ, Zhu J, Kopcic E, Post IJM, et al. Detailed analysis of an enriched deep intronic ABCA4 variant in Irish stargardt disease patients. Sci Rep. 2023;13(1):9380.
Ahmad A, Strohbuecker S, Tufarelli C, Sottile V. Expression of a SOX1 overlap** transcript in neural differentiation and cancer models. Cell Mol Life Sci. 2017;74(22):4245–58.
da Fontoura CS, Miller SF, Wehby GL, Amendt BA, Holton NE, Southard TE, et al. Candidate gene analyses of skeletal variation in malocclusion. J Dent Res. 2015;94(7):913–20.
Zawislak A, Wozniak K, Agirre X, Gupta S, Kawala B, Znamirowska-Bajowska A et al. Association of ABCA4 gene polymorphisms with cleft lip with or without cleft palate in the polish population. Int J Environ Res Public Health. 2021;18(21).
Yuan Q, Blanton SH, Hecht JT. Association of ABCA4 and MAFB with non-syndromic cleft lip with or without cleft palate. Am J Med Genet A. 2011;155A(6):1469–71.
Neela PK, Gosla SR, Husain A, Mohan V, Thumoju S, Bv R. Association of MAPK4 and SOX1-OT gene polymorphisms with cleft lip palate in multiplex families: a genetic study. J Dent Res Dent Clin Dent Prospects. 2020;14(2):93–6.
Yildirim M, Seymen F, Deeley K, Cooper ME, Vieira AR. Defining predictors of cleft lip and palate risk. J Dent Res. 2012;91(6):556–61.
Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42(6):525–9.
Al-Zaid FS, Hurley MJ, Dexter DT, Gillies GE. Neuroprotective role for RORA in Parkinson’s disease revealed by analysis of post-mortem brain and a dopaminergic cell line. NPJ Parkinsons Dis. 2023;9(1):119.
Li M, Olotu J, Buxo-Martinez CJ, Mossey PA, Anand D, Busch T, et al. Variant analyses of candidate genes in orofacial clefts in multi-ethnic populations. Oral Dis. 2022;28(7):1921–35.
Reyes-Gibby CC, Wang J, Yeung SJ, Chaftari P, Yu RK, Hanna EY, et al. Genome-wide association study identifies genes associated with neuropathy in patients with head and neck cancer. Sci Rep. 2018;8(1):8789.
Urraca N, Hope K, Victor AK, Belgard TG, Memon R, Goorha S, et al. Significant transcriptional changes in 15q duplication but not Angelman syndrome deletion stem cell-derived neurons. Mol Autism. 2018;9:6.
Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, et al. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6(4):268–79.
Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004.
Acknowledgements
Sincere thanks to the research participants of this project.
Funding
Support was provided by NIH Grant R21-DE16718 and R01-DE14899. Genoty** services were provided by the Center for Inherited Disease Research (CIDR). The CIDR is fully funded through a Federal contract from the National Institutes of Health to The Johns Hopkins University (contract number N01-HG-65403).
Author information
Authors and Affiliations
Contributions
MB – Conceptualization, investigation, methodology, and writing original draft. CC – Investigation, methodology, software, writing, review, and editing. ARV – Conceptualization, investigation, obtained funding, review, and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from all participants and this protocol has both the University of Pittsburgh and local Filipino IRB approval.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bezamat, M., Carver, C.E. & Vieira, A.R. Family-based GWAS for dental class I malocclusion and clefts. BMC Oral Health 24, 665 (2024). https://doi.org/10.1186/s12903-024-04444-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-024-04444-x