
Abstract
Background
Despite the growing demand for antimicrobial peptides (AMPs) for clinical use as an alternative approach against antibiotic-resistant bacteria, the manufacture of AMPs relies on expensive, small-scale chemical methods. The small ubiquitin-related modifier (SUMO) tag is industrially practical for increasing the yield of recombinant proteins by increasing solubility and preventing degradation in expression systems.
Results
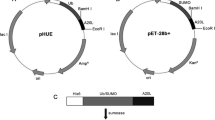
A new vector system, pKSEC1, was designed to produce AMPs, which can work in prokaryotic systems such as Escherichia coli and plant chloroplasts. 6xHis was tagged to SUMO for purification of SUMO-fused AMPs. Abaecin, a 34-aa-long antimicrobial peptide from honeybees, was expressed in a fusion form to 6xHis-SUMO in a new vector system to evaluate the prokaryotic expression platform of the antimicrobial peptides. The fusion sequences were codon-optimized in three different combinations and expressed in E. coli. The combination of the native SUMO sequence with codon-optimized abaecin showed the highest expression level among the three combinations, and most of the expressed fusion proteins were detected in soluble fractions. Cleavage of the SUMO tag by sumoase produced a 29-aa-long abaecin derivative with a C-terminal deletion. However, this abaecin derivative still retained the binding sequence for its target protein, DnaK. Antibacterial activity of the 29-aa long abaecin was tested against Bacillus subtilis alone or in combination with cecropin B. The combined treatment of the abaecin derivative and cecropin B showed bacteriolytic activity 2 to 3 times greater than that of abaecin alone.
Conclusions
Using a SUMO-tag with an appropriate codon-optimization strategy could be an approach for the production of antimicrobial peptides in E.coli without affecting the viability of the host cell.
Similar content being viewed by others

Background
The overuse of antibiotics for the last several decades and the prevalence of antibiotic-resistant bacterial infections present a threat to global health. About 30 million people are projected to be killed by antibiotic-resistance bacteria by 2050 [1]. Therefore, the discovery of novel antibiotic agents is of increasing medical importance. However, there is also a potential that over-use or abuse of a new antibiotic will give rise to more antibiotic-resistance bacteria. Growing difficulties from the discovery of new antibiotics to their clinical use have diverted attentions to a new therapeutic agent, antimicrobial peptides (AMPs) [2].
As an alternative approach to prevent the spread of bacteria which are resistant to antibiotics, antimicrobial peptides (AMPs) have been extensively studied since they have a broad spectrum of anti-infective activity against pathogenic bacteria with relatively low minimal inhibitory concentrations and with a property less capable of incurring resistance than conventional antibiotics, due to their nonspecific interaction with bacterial membranes, and their ability to work on multiple targets. In addition to the direct antimicrobial activities, many AMPs are associated with immunomodulatory properties as seen in host-defense peptides [3, 4].
To meet the demand of AMPs for clinical applications, the produciton of AMPs can be achieved by chemical synthesis. For example, phase peptide synthesis approaches can provide easy isolation of peptides with high purity and less use of solvents by avoiding chromatographic purification [5]. However, chemical synthesis has several disadvantages that prevent cost-effective, industrial-scale production of AMPs. In addition, the synthesis of longer peptides with more than 50 amino acids is not favored [6].
For high production of large peptides, biological systems such as bacteria and yeast have been preferably used as production platforms [7,8,9,10,11,12,13]. Although these biological systems don’t need expensive active pharmaceutical ingredients (APIs) and toxic chemical solvents, there are still some issues remaining, such as the toxicity of the expressed AMPs to host cells, expensive purification, and low yield. As a solution to address these issues, plants can be a viable alternative expression platform [14,Full size image
The vector was further modified by the addition of tagging systems. SUMO, derived from human SUMO1, was attached to abaecin to increase solubility and prevent toxicity of abaecin to host cells. Also, a 6xHis purification tag was added to the N-terminus of SUMO to facilitate the purification of the SUMO-fused abaecin, and to isolate abaecin from SUMO after cleavage by sumoase. Three different combinations of codon-optimized synthetic sequences of the 6xHisSUMO-abaecin were cloned into the new vector, and their expression in an E. coli system was investigated after the confirmation of their sequences (Fig. 1a, b and Additional file 1: Figure S1). For the codon optimization, codon adjustment was performed according to a previous study [33], in which a new algorithm for codon optimization was developed based on the codon usage hierarchy of chloroplast psbA genes, and the codon-optimized sequences under the control of psbA/5’ UTR showed increased expression levels over their respective native sequences in both E. coli and plant chloroplasts. The expressions of all the three fusion constructs didn’t show any toxicity to host cells. As shown in Fig. 1c, there was no significant difference of optical density (OD) values between untransformed and transformed cells with the three fusion constructs, which were grown overnight at 37 °C.
Evaluation of expression of His-tagged SUMO fused abaecin in E. coli
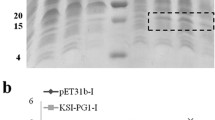
The three constructions of the 6xHisSUMO-abaecin fusion protein were evaluated using an E. coli expression system. Transformed E. coli cells with the three constructs were grown in liquid culture and the relative expression levels between the three fusion proteins were examined using an immunoblot assay with anti-His antibody. The expressed fusion proteins were detected around 20 kDa, not at 15.7 kDa which is a deduced molecular weight (Fig. 2a). This kind of discrepancy is very often observed in proline-rich proteins expressed in E. coli, due to presumably increased rigidity caused by high proline content, leading to slower migration than the same molecular weight protein [38,39,40,41]. Abaecin (3.9 kDa, 34 amino acids) contains 10 prolines in total of 34 amino acids, accounting for 34.9%, so it is assumed that the high content of proline residues affected the migration of all three fusion proteins (136 amino acids in length for each construct). Among three fusion proteins, 6xHisSUMO (N)-abaecin (C) (N and C, stand for “native sequence” and “codon-optimized sequence”, respectively) showed the highest expression, with 2.8 or 3.5 fold higher expression than 6xHisSUMO(C)-abaecin (N) or 6xHisSUMO(C)-abaecin(C), respectively (Fig. 2b). All three constructs showed that the expressed fusion proteins were detected at levels 1.4 to 2.1 times higher in the soluble fraction than in the insoluble one (Fig. 2b).

Western blot analysis for the comparison of expression levels between three different codon-optimized 6xHisSUMO-abaecin DNA constructs in E.coli, sumoase assay, and purification. a Comparison of the level of expression between three different combinations of 6xHisSUMO-abaecin fusion protein using western blot. H6SU-Abe, 6xHisSUMO-abaecin; N, native sequence; C, codon-optimized sequence; S, soluble fraction; I, insoluble fraction; DH5α, untransformed E. coli; M, protein molecular size marker. The fusion proteins were detected using anti-His antibody. Each lane was loaded with 20 μg of protein. b Comparison of band intensities detected in (a). The band intensities from 5 independent western blot images were extrapolated using ImageJ software and represented with standard deviation. c Cleavage assay of the 6xHisSUMO (N)-abaecin (C) by sumoase. Coomassie staining assay (left panel) to detect cleavage products. Western blot assay (right panel) to investigate the cleavage efficacy of SUMO from 6xHisSUMO-abaecin by sumoase. *, 6xHisSUMO-abaecin; **, cleaved 6xHisSUMO; ***, cleaved abaecin; −, no treatment of sumoase; +, 6 h treatment of sumoase. Each lane was loaded with 20 μg of protein. d Western blot assay for the purified 6xHisSUMO-abaecin from E. coli using gravity Ni column. T, total protein; FT, flow-through; W, wash; E, elution
From these data, we confirmed that the newly designed vector is operable in a prokaryotic system and that the native sequence of the heterologous human SUMO gene improved the expression of the SUMO-fused abaecin in E. coli (Fig. 2a).
Cleavage of abaecin from purified His-tagged SUMO-abaecin and MALDI-TOF analysis
The cleavage efficiency of sumoase on the fusion protein was investigated by treating the soluble proteins extracted from E. coli with sumoase for 6 h and then detecting the treated proteins with Coomassie staining and western blot. As seen in Fig. 2c, the fusion proteins detected at 19 kDa before treatment of sumoase were cleaved and produced two products (Fig. 2c). To confirm the result, an immunoblot assay with a replicate gel was performed using anti-His antibody. The band for the cleaved-off 6xHisSUMO domain was detected around 16 kDa, along with a band for the fusion protein around 19 kDa (Fig. 2c). This data confirmed that the tertiary structure of SUMO was correctly formed in E. coli and recognized by sumoase (Fig. 2c).
With confirmation of the proper expression of 6xHisSUMO-abaecin and cleavage of SUMO tag (Fig. 2c), the fusion proteins were purified for mass analysis of abaecin cleaved from the fusion protein (Fig. 2d). The abaecin cleaved by sumoase was examined by MALDI-TOF analysis to verify the precise cleavage between SUMO and abaecin. Singly protonated molecular ion (M + H) + of the cleaved-off abaecin was observed at m/z 3245.034 Da using reflector mode (Fig. 3a), while the theoretical molecular mass is 3877.049 Da (Fig. 3b). This discrepancy between the observed and theoretical values suggests the possibility of deletion of abaecin peptide at either N- or C- termini. To identify the peptide peak, we calculated the masses for a series of deleted abaecin peptides from either N- or C-termini and compared the theoretical masses to the observed mass (Fig. 3b) The best amino acid sequence corresponding to the observed mass at 3245.034 Da was deduced as YVPLPNVPQPGRRPFPTFPGQGPFNPKIK, which indicates the deletion of 5 amino acids from C-terminus of abaecin (Fig. 3b). Based on the results, although the proper cleavage between SUMO and abaecin occurred, undesired cleavage happened at the C-terminus.

MS analysis of the cleaved-off abaecin using MALDI-TOF and investigation of binding sequence of 29-aa-long abaecin to DanK. a MS spectra obtained from reflector mode. The reflector mode was performed using α-cyano-4-hydroxycinnamic acid (CHCA) matrix for the lower range of mass from 0.6 to 6 kDa to analyze the target peak at high resolution. b Table of masses and overall charge for a series of deleted peptide sequences from abaecin. Masses were analyzed for each deleted peptide sequence from either N- or C-termini in order to find the best peptide sequence matchable to the detected mass (m/z), 3245.034. Net charges at pH 7 were calculated by Peptide Property Calculator (https://www.biosyn.com/peptidepropertycalculatorlanding.aspx).The amino acids which affect the overall charge of abaecin were represented as bold fonts, such as K for Lys and R for Arg. c Sequence analysis for the binding affinity of the 29-aa-long abaecin to DnaK. The amino acid sequence of the 29-aa-long abaecin is represented, and the putative binding sequence to DnaK is underlined and numbered
Since the small cationic peptides are very susceptible to proteolytic degradation in E.coli [10], the positive charge distribution of the abaecin was analyzed (Fig. 3b) from which the positive charge patch created by two Lys (K) residues at C-terminus was likely targeted by proteolytic attack.
Next, we analyzed whether or not the 29-aa-long abaecin still retained the functional binding sequence to its intracellular target molecule, DnaK. For the analysis, we used limbo server (http://limbo.switchlab.org) [42] and found that the shorter form of abaecin still had a binding sequence to the DnaK; YVPLPNV, numbered from 1 to 7 (score 1.65) (Fig. 3c). Therefore, we continued to test an antimicrobial activity of the 29-aa-long abaecin derivative with or without cecropin B against Bacillus subtilis.
Antibacterial activity of purified abaecin against Bacillus subtilis
To evaluate the antimicrobial activity of 29-aa abaecin expressed in E. coli, the activity was tested against B. subtilis using an agar diffusion assay with the purified abaecin derivative. We also tested whether the SUMO-fused abaecin is toxic to bacteria when treated externally, although we had already confirmed that there was no toxicity of the 6xHisSUMO-fused abaecin to host cells when expressed in the cells (Fig. 1c). The treatment of the 6xHisSUMO-abaecin didn’t show bacteriolytic activity against B. subtilis (Fig. 4a, b). In the preliminary test, we found that anti-B. subtilis activity of 29-aa abaecin required an amount of 1.7 μg (Fig. 4a, b). The antimicrobial activity of 29-aa abaecin was enhanced in a combination treatment with cecropin B. No antimicrobial activity was observed by cecropin B when 0.125 μg of the peptide was treated (Fig. 4a, b), however, the antimicrobial activity of abaecin, at the dose of 1.7 μg, was increased by 40% when combined with the cecropin B (0.125 μg) (Fig. 4a, b). The antibacterial activity of abaecin was further enhanced by the increase of cecropin B quantity up to 0.25 μg (Fig. 4a, b). These results are consistent with the previous reports that the functional interaction of abaecin with other pore-forming peptides, such as cecropin A, stomoxyn and hymenoptaecin, at their sublethal concentrations, reciprocally potentiates bacteriolytic activity by increasing membrane permeabilization [13, 31, 43].

Evaluation of antibacterial activity of 29-aa-abaecin with or without cecropin B. a The antibacterial activities of 29-aa abaecin with or without cecropin B against B. subtilis. Agar plates spread with 100 μl of B. subtilis liquid culture grown overnight were punctured and dropped with 10 μl of abaecin or cecropin B or both peptides, which were purified and resuspended in 1X PBS, then incubated at 37 °C for 16 h. The antimicrobial peptides and their amount treated into the corresponding wells are represented in (b). b Evaluation of inhibitory effect by antimicrobial peptides on B. subtilis by measuring the inhibited zone area. The inhibited zone areas of B. subtilis were extrapolated using ImageJ software. Scale bars represent 1 cm
Taken all together, 6xHisSUMO-tagged abaecin expressed in a soluble form in E. coli caused no toxicity to the host cells, and the tag was properly recognized and removed by sumoase, but the cleaved abaecin was 29-aa long in size, not 34-aa long. However, the purified 29-aa-long abaecin retained its antimicrobial activity and the activity was potentiated by co-treatment with a pore-forming antimicrobial peptide, cecropin B.
