
Abstract
NUDT2 is an enzyme important for maintaining the intracellular level of the diadenosine tetraphosphate (Ap4A). Bi-allelic loss of function variants in NUDT2 has recently been reported as a rare cause of intellectual disability (ID). Herein, we describe a Chinese girl with ID, attention deficit hyperactivity disorder (ADHD), and motor delays with abnormal walking posture and difficulty climbing stairs, who bears compound heterozygous variants c.34 C > T (p.R12*) and c.194T > G (p.I65R) in NUDT2. Homozygous variants c.34 C > T (p.R12*) or c.186del (p.A63Qfs*3) in NUDT2 were previously reported to cause ID. This is the first patient with ID due to compound heterozygous variants in NUDT2 and p.I65R is a novel missense variant. This study enriched the genotype and phenotype of NUDT2-related ID and supported the critical developmental involvement of NUDT2.
Similar content being viewed by others


Introduction
Global developmental delay (GDD) and intellectual disability (ID) are phenotypically and genetically heterogeneous with common morbidity affecting at least 2 ~ 3% of the population [1]. Investigation for genetic causes of GDD/ID has contributed to the identification of hundreds of disease-causing genes. However, challenges remain in the specific diagnosis, as there is significant variability in the clinical features. Trio-based whole-exome sequencing (Trio-WES) analysis gives a high diagnostic yield of over 50% in children with severe developmental delay if used as a first-tier diagnostic test [2]. NUDT2 (Nudix hydroxylase 2; Nudix-type motif 2) is a member of the MutT family of nucleotide pyrophosphatases, which catalyzes the asymmetric hydrolysis of Ap4A to yield AMP and ATP. NUDT2 also exhibits decap** activity towards FAD or dpCoA-capped RNAs in vitro and involves viral RNA degradation and antiviral immunity [3, 4]. This gene may be a prognostic marker in breast carcinomas [5,6,7]. So far, 11 GDD/ID patients have been reported exhibiting homozygous variants in NUDT2, suggesting a neurodevelopmental role [8,9,10,11]. Here, we performed Trio-WES and identified novel compound heterozygosity for NUDT2 variants in a Chinese patient, affected with GDD/ID, but also with ADHD, further expanding the genotypic and phenotypic spectrum associated with this gene.
Materials and methods
Whole-exome sequencing
Trio-WES and Sanger sequencing was performed on genomic DNA extracted from the peripheral blood from the proband and her parents. Libraries were prepared using xGen Exome Research Panel v1.0 (IDT, Iowa, USA) followed by sequencing on the Illumina NovaSeq 6000 (Illumina San Diego, CA). Sequences underwent processing, including adapter trimming and filtering of low-quality reads using fastp, alignment to the GRCh37/hg19 reference genome was performed with Burrows-Wheeler Aligner (BWA), and variant calling of single nucleotide variants (SNVs) as well as insertions and deletions < 50 bp (small Indels) was executed by Genome Analysis Tool Kit (GATK) [12,13,14]. Basic filtering excluded the following variants: (1) Wild-type in the proband, (2) Intronic variants deeper than 30 bp, (3) Low-quality SNPs (variant frequency < 0.2 or sequencing depth < 4X or quality value < 35), (4) Low-frequency simple tandem repeat Indels (SSR > 7 and AF < 0.3), (5) Indels larger than 50 bp. Annotation of high-quality variants utilized dbSNP, gnomAD, 1000 Genomes Project, ExAC, ESP, and Chigene in-house MAFs database. Additional databases such as OMIM, HGMD, and ClinVar were used. Annotation was performed using a variant annotation software independently developed by Bei**g Chigene Translational Medicine Research Center Co., Ltd, 100,875, Bei**g. Computational prediction tools, including Provean, SIFT, Polypen2, MutationTaster, M-Cap, and REVEL software packages, were employed to assess the potential pathogenicity of missense variants. MaxEntScan, dbscSNV, and GTAG software packages were used to predict the functional impact of variants on splicing sites. Variant prioritization focused on rare variants with moderate or high predicted impacts, such as frameshift, nonsense, splicing, and missense. Phenotype consistency was evaluated through Human Phenotype Ontology (HPO) terms. Trio analysis assessed de novo, compound heterozygous, and recessive models. Top candidate variants underwent manual review, considering IGV visualization, inheritance patterns, gene function, and patient phenotype. WES identified potential pathogenic variants aligned with the patient’s phenotype and family history. All variants were assessed following the American College of Medical Genetics and Genomics (ACMG) guidelines [15]. The identified variants and segregation analysis were confirmed by Sanger sequencing using specific primers.
Results
Clinical description
The proband was a 5-year-old female to healthy non-consanguineous parents of Chinese ethnicity. She was born at week 40+ 5 by caesarian section with a birth weight of over 3 kg and a length of 52 cm. No history of neonatal complications or feeding difficulties was observed. Milestones of motor development were delayed as she sat at 9 months, stood at 2 years, and walked unsupported at 2 years 4 months. For her speech, she vocalized “mama, baba” at 10 months and began using single words at 1 year. On current examination at 6 years, her growth parameters weight (30 kg, 97th centile), and height (123 cm, 90th centile) were within normal range. She had subtle facial dysmorphism, such as slightly upslanting palpebral fissures. Her language expression lagged behind that of other children of the same age and slightly poor logic of speech with unclear articulation. Testing with the Wechsler Intelligence Scale revealed 84 for the verbal IQ, 63 for the performance IQ, 71 for the full-scale IQ, showing cognitive skills mildly impaired. She presented with ADHD and deficits in executive function, including slow processing speed, poor learning, and working memory performance. Muscular hypotonia in all extremities and motor impairments with an unsteady gait, climbing stairs difficulties were also noted. Brain magnetic resonance imaging (MRI) revealed thinning but fully formed corpus callosum (Fig. 1A), without other abnormalities. Electroencephalogram (EEG) was normal. Electromyography (EMG) examination demonstrated sensorimotor neurogenic lesions in the distal lower extremities with axonal features in a length-dependent manner, and a normal nerve conduction velocity and SNAP amplitudes.
Molecular analysis
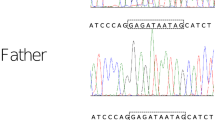
Trio-WES revealed the proband harboring compound heterozygous variants at NUDT2 NM_001244390: c.34 C > T (p.R12*) and c.194T > G (p.I65R), which were inherited from his mother and father, respectively (Fig. 1B). Sanger sequencing further confirmed both asymptomatic parents to be heterozygous carriers, following the rule of autosomal recessive inheritance (Fig. 1C). Following the ACMG guidelines, the c.34 C > T (NM_001244390) variant in exon 2 is classified as pathogenic, documented in dbSNP with rs number rs148119952. The variant has a frequency of 0.0024 in the 1000 Genomes Project and is absent in gnomAD. It is a nonsense mutation (PVS1), expected to result in premature protein termination. The pathogenic p.R12* has been reported as a homozygous LOF variant in seven GDD/ID patients (PM3) and showed genotype-phenotype segregation in one family (PP1-Moderate). The missense variant of uncertain significance (VUS) p.I65R is absent in the population databases such as 1000 genomes, ExAC, ESP and gnomAD or our internal database of exomes. So it is considered a rare variant although this variant has been recorded in the dbSNP database with the rs number rs765777597 (PM2). Various protein damage prediction software tools, including MutationTaster and SIFT, suggest that this variant is deleterious (PP3). Of the six missense variants in ClinVar, c.174G > T (p.Glu58Asp) was likely pathogenic in intellectual developmental disorder with or without peripheral neuropathy (autosomal recessive inheritance), submitted by the University Hospital Tübingen. Furthermore, residue I65 is positioned near the Nudix box domain of NUDT2 and is highly evolutionary conserved across species (Fig. 2A and B). In molecular modeling, I65 localizes in the loop between the first and second helixes, if it mutates to R65, which replaces hydrophobic isoleucine with a basic polar arginine, will generate unfavorable torsion angles and lead to a change in protein thermodynamic stability (ΔΔG) and thus destabilize the structure of NUDT2, indicating a likely impact on the protein function (Fig. 2C). No other rare variants were identified by Trio-WES in the patient that could explain the observed phenotype. With her clinical profile, especially the presence of ID and delayed motor development resembling the reported cases’ clinical presentations, we considered the identified NUDT2 variants as the likely cause explaining our patients’ disease phenotypes.

A Chinese family with NUDT2 compound heterozygous variants causing intellectual disability. (A) Sagittal brain MRI of the patient. Note thinning of the corpus callosum (arrow). (B) Pedigree of the family. An arrow indicates the proband. The patient (II-1) had compound heterozygous variants p.I65R and p.R12*, which were inherited from her father (I-1) and mother (I-2), respectively. (C) Sanger sequencing pherograms show compound heterozygosity of NUDT2 in the proband and heterozygosity in both parents

Identification of a novel NUDT2 missense variant. (A) Schematic representation of NUDT2 transcript (NM_001244390) and protein (NP_001231319) with the light blue box delineates the catalytically active Nudix Box. Previously identified variants in GDD/ID are indicated in blue. The identified novel variant is indicated in red. The secondary structure of NUDT2 protein (six beta-strands and four helixes) shows I65 located at the loop between the first and second helixes. (B) Conservation of the identified variants in NUDT2. The residue R12 and I65 (marked with red arrows) are highly conserved among species. (C) Location of I65R mutant in NUDT2 protein (AlphaFold: AF-P50583-F1) and the fold stability change prediction (ΔΔG) upon I65R mutant using four methods (mCSM/SDM/DUET/CUPSAT). I65R destabilizes overall stability
Discussion
NUDT2, located at position 9p13.3, contains 3 exons and encodes a 147-amino acid Ap4A hydrolase containing a MutT motif or“nudix”. The protein is believed to play a major role in maintaining the low level of intracellular Ap4A, the function of which has yet to be established [6]. NUDT2-related ID is a rare entity. The variants identified in NUDT2 so far included one nonsense and one frameshift variant that exert their effect through loss-of-function (LOF) [8,9,10,11]. Thus, our knowledge regarding NUDT2 defects that lead to clinical manifestations of GDD/ID is limited. The present study describes a 5 years-old female from a Chinese family with ID, who bears one known nonsense variant and a newly described missense variant, supporting the previous findings and expanding the mutational spectrum for autosomal recessive ID.
Until now, only 11 reported cases with neurological symptoms due to two variants in the NUDT2 gene have been described in 7 pedigrees in homozygous conditions (Table 1). Most cases were in consanguine families of Saudi Arabian origin (7 cases, 64%), two cases were of Mexican descent and one case was of Cajun descent. NUDT2-related disorders included ID and GDD. Additional signs comprised hypotonia, delayed motor and language development, and cognitive impairment; and less frequently, ataxia, leukodystrophy, thinning of the corpus callosum, denervation atrophy, demyelination, or axonal sensorimotor polyneuropathy. Non-neurological features encompassed low birth weight and height, neonatal feeding problems, subtle facial dysmorphism, and microcephaly. The first and second studies described seven cases with homozygous p.R12* in NUDT2 to underlie ID/GDD [8, 9]. The four patients presented in the third and fourth studies with homozygous p.A63Qfs*3 in NUDT2 also developed polyneuropathy in addition to ID [10, 11]. We report the first case cosegregated with compound heterozygous mutations (p.R12* and p.I65R) in NUDT2. Although our case presented some of the recognized features, ADHD is first described here, and low birth weight and height, weak sucking in infancy, and microcephaly were not observed in our patient. Our case also showed a length-dependent axonal sensorimotor polyneuropathy, which suggested that progressive sensorimotor neuropathy may be invariably present. The two affected sisters with homozygous p.R12*, walked by 4 years of age and vocalized “mama, baba” at 2.5 years [8]. The three patients with homozygous p.A63Qfs*3 walked at the age of 3 and began using single words at age 2 [10]. In contrast, our patient walked unsupported at 2 years 4 months. Her parents noticed no delays in language development but recently noted a decrease in speech articulation. Truncating variant p.R12* at N-terminal is predicted to trigger nonsense-mediated decay (NMD) and impair the enzymatic domain and p.A63Qfs*3 in the last exon is unlikely to undergo NMD. The missense mutation p.I65R reported here resides in a conserved region near the Nudix box domain but may not define pathogenic LOF alleles, thereby differential activities of the protein placing NUDT2-related disease towards the milder phenotypes of neurodevelopmental disorders.
In conclusion, this study contributed further to the characterization of NUDT2-related disorders and identified novel compound heterozygosity as the cause of disease, allowing for accurate genetic counseling. Our results supported that variants in the NUDT2 cause a multisystem disease with intellectual disability and polyneuropathy, and more research is needed to study the underlying mechanisms of NUDT2-related disorders and the genotype-phenotype correlations. The clinical possibility of NUDT2 biallelic mutation should be considered in children with GDD/ID.
Data availability
All data generated or analysed during this study are included in this published article. The original contributions presented in this study are publicly available. The NUDT2 variants NM_001161.4: c.34 C > T/p.Arg12*, c.194T > G/p.Ile65Arg were submitted to the LOVD database (https://databases.lovd.nl/shared/transcripts/NUDT2), with the LOVD Variant ID: https://databases.lovd.nl/shared/variants/0000932973#00014895, https://databases.lovd.nl/shared/variants/0000932972#00014895.
References
Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond). 2017;17(6):558–61. https://doi.org/10.7861/clinmedicine.17-6-558.
Bass N, Skuse D. Genetic testing in children and adolescents with intellectual disability. Curr Opin Psychiatry. 2018;31(6):490–5. https://doi.org/10.1097/YCO.0000000000000456.
Laudenbach BT, Krey K, Emslander Q, et al. NUDT2 initiates viral RNA degradation by removal of 5’-phosphates. Nat Commun. 2021;12(1):6918. https://doi.org/10.1038/s41467-021-27239-y.
Sharma S, Grudzien-Nogalska E, Hamilton K, et al. Mammalian nudix proteins cleave nucleotide metabolite caps on RNAs. Nucleic Acids Res. 2020;48(12):6788–98. https://doi.org/10.1093/nar/gkaa402.
Kwon O, Kwak D, Ha SH, et al. Nudix-type motif 2 contributes to cancer proliferation through the regulation of rag GTPase-mediated mammalian target of rapamycin complex 1 localization. Cell Signal. 2017;32:24–35. https://doi.org/10.1016/j.cellsig.2017.01.015.
Marriott AS, Vasieva O, Fang Y, Copeland NA, McLennan AG, Jones NJ. NUDT2 disruption elevates Diadenosine Tetraphosphate (Ap4A) and down-regulates Immune Response and Cancer Promotion genes. PLoS ONE. 2016;11(5):e0154674. https://doi.org/10.1371/journal.pone.0154674.
Oka K, Suzuki T, Onodera Y, et al. Nudix-type motif 2 in human breast carcinoma: a potent prognostic factor associated with cell proliferation. Int J Cancer. 2011;128(8):1770–82. https://doi.org/10.1002/ijc.25505.
Anazi S, Maddirevula S, Faqeih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22(4):615–24. https://doi.org/10.1038/mp.2016.113.
Yavuz H, Bertoli-Avella AM, Alfadhel M, et al. A founder nonsense variant in NUDT2 causes a recessive neurodevelopmental disorder in Saudi arab children. Clin Genet. 2018;94(3–4):393–5. https://doi.org/10.1111/cge.13386.
Diaz F, Khosa S, Niyazov D, et al. Novel NUDT2 variant causes intellectual disability and polyneuropathy. Ann Clin Transl Neurol. 2020;7(11):2320–5. https://doi.org/10.1002/acn3.51209.
Lee H, Huang AY, Wang LK, et al. Diagnostic utility of transcriptome sequencing for rare mendelian diseases. Genet Med. 2020;22(3):490–9. https://doi.org/10.1038/s41436-019-0672-1.
MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–76. https://doi.org/10.1038/nature13127.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. https://doi.org/10.1093/bioinformatics/btp698.
McKenna A, Hanna M, Banks E, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Acknowledgements
We are grateful to the patient and parents who supported our study.
Funding
This work was supported by grants from the Science Foundation of Wuhan Health and Family Planning Commission (grant number WZ22Q33 to Yangcan Ming). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Bo Bi, **aohong Chen and Shan Huang wrote the main manuscript text. Min Peng performed the molecular analysis and interpreted the genetic data. Weiyue Gu prepared Figs. 1 and 2.Hongmin Zhu and Yangcan Ming conceived and designed the experiments, prepared figures and table, authored drafts of the article.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The patient was admitted to the rehabilitation department of Wuhan Children’s Hospital in September 2021. All procedures performed in studies involving human participants were conformed to the ethical guidelines of the 1975 Declaration of Helsinki and approved by the Ethical Committee of Wuhan Children’s Hospital (Ethical approval number: 2022R043-E01). Written informed consent for genetic analysis was obtained from the parents of the affected individual.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bi, B., Chen, X., Huang, S. et al. The first case of intellectual disability caused by novel compound heterozygosity for NUDT2 variants. BMC Pediatr 24, 60 (2024). https://doi.org/10.1186/s12887-024-04542-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04542-3