
Abstract
Background
Congenital cataract-microcornea syndrome (CCMC) is characterized by the association of congenital cataract and microcornea without any other systemic anomaly or dysmorphism. Although several causative genes have been reported in patients with CCMC, the genetic etiology of CCMC is yet to be clearly understood.
Purpose
To unravel the genetic cause of autosomal dominant family with CCMC.
Methods
All patients and available family members underwent a comprehensive ophthalmologic clinical examination in the hospital by expert ophthalmologists and carried out to clinically diagnosis. All the patients were screened by whole-exome sequencing and then validated using co-segregation by Sanger sequencing.
Results
Four CCMC patients from a Chinese family and five unaffected family members were enrolled in this study. Using whole-exome sequencing, a missense mutation c.295G > T (p.A99S, NM_003106.4) in the SOX2 gene was identified and validated by segregation analysis. In addition, this missense mutation was predicted to be damaging by multiple predictive tools. Variant p.Ala99Ser was located in a conservation high mobility group (HMG)-box domain in SOX2 protein, with a potential pathogenic impact of p.Ala99Ser on protein level.
Conclusions
A novel missense mutation (c.295G > T, p.Ala99Ser) in the SOX2 gene was found in this Han Chinese family with congenital cataract and microcornea. Our study determined that mutations in SOX2 were associated with CCMC, warranting further investigations on the pathogenesis of this disorder. This result expands the mutation spectrum of SOX2 and provides useful information to study the molecular pathogenesis of CCMC.
Similar content being viewed by others
Introduction
Congenital cataract is one of the leading causes of childhood blindness [1]. The overall prevalence of congenital cataract has been reported to be 0.63 to 9.74 per 10,000 (median = 1.71) [2]. Moreover, non-syndromic congenital cataracts may occur alone or accompanied by various congenital ocular anomalies, covering from aniridia, microcornea, microphthalmia to neurological and renal diseases [3, 4]. The high genetic heterogeneity of congenital cataracts is a challenge for establishing a reliable genotype-phenotype correlation in the clinical setting [5]. Congenital cataract-microcornea syndrome (CCMC; OMIM 116200) is marked genetic heterogeneity and the association of congenital cataract and microcornea (a horizontal corneal diameter of less than 10.00 mm) without any other systemic anomaly or dysmorphism [6], appears as a distinct phenotype affecting 12-18% of heritable congenital cataract patients [7]. The molecular basis of cataract with microcornea in the absence of microphthalmia, anterior segment dysgenesis, or coloboma has not been elucidated.
Next-Generation Sequencing (NGS), also known as high-throughput sequencing, has been increasingly applied to congenital cataract [4, 8]. To date, genetic studies have identified mutations from more than 87 causative genes in non-systemic congenital cataract, and genetic mutations remain the leading cause of congenital cataracts (data from Cat-Map http://cat-map.wustl.edu/) [9]. Among these genes, mutations in at least 19 genes and loci have been reported to be responsible for congenital cataract with microcornea, including (EPHA2, FOXE3, GJA8, CRYGD, CRYGC, MAB21L2, ATOH7, SLC16A12, BEST1, ARL2, MIP, GJA3, MAF, CRYAA, CRYBB3, CRYBB2, CRYBB1, CRYBA4, NHS) [9].
This study identified a novel heterozygous missense mutation in the SOX2 gene by whole-exome sequencing in an autosomal dominant CCMC family. This mutation was not observed in any of the healthy family members. With WES technologies, our study expands the spectrum and provides additional genotype-phenotype associations in CCMC.
Materials and methods
Family ascertainment and DNA specimens
This study was approved by the Ethical Committee of the Eye Hospital of Wenzhou Medical University and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each participant. All patients and available family members underwent a comprehensive ophthalmologic clinical examination by expert ophthalmologists and carried out to clinically diagnose. Peripheral blood samples were collected from patients and available family members. A conventional DNA extraction kit (Simgen, Hangzhou, China) was used for genomic DNA extraction. In addition, blood samples from another 200 healthy controls without of eye disease or syndromes from the Han Chinese were collected.
Whole-exome sequencing and analysis
DNA samples from affected individuals were subjected to whole-exome sequencing (WES). WES was performed using IDT’s xGen Exome Research Panel V1.0 (Integrated DNA Technologies, San Diego, USA) and Novaseq6000 platform (Illumina, San Diego, USA) for sequencing. Human reference genome (hg19/GRCh37) was used as a reference genome to align sequence reads. Protocols for next-generation sequencing and data analysis, including copy number variation analysis, have been recently published [4, 10]. Briefly, variants were filtered for rare or absent variants in the 1000 Genomes Project database (http://www.1000genomes.org/) and minor allele frequency (MAF) less than 0.01 in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/).
In silico analysis
In the case of missense variants, six different predictive software were applied to estimate functional effects, including SIFT (Sorting Intolerant From Tolerant) [11], MAPP (Multivariate Analysis of Protein Polymorphism) [12], MutationTaster2 [12], PolyPhen2 [13], CADD (Combined Annotation Dependent Depletion) score [12], and REVEL (Rare Exome Variant Ensemble Learner) [14]. Variants were considered if estimated to be disease-causing by at least 2 of 6 algorithms.
Variant confirmation and segregation analysis
Sanger sequencing was used to validate variants, as well as for segregation analysis. Sanger sequencing was then performed in 200 unrelated controls for mutation validation and prevalence testing. The topological model of the SOX2 polypeptide was shown [15], and crystal structures of the wild-type and mutant proteins were predicted with Phyre2 [16] and visualized with PyMol software (Version 1.5).
Results
Clinical findings
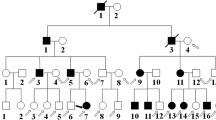
A three-generation Chinese pedigree that consists of 9 individuals, including four affected individuals, provided the basis for the study (Fig. 1). The autosomal dominant pattern of inheritance was ascertained from the pedigree (Fig. 1). Clinical data of affected individuals are given in Table 1. The proband was a 46-year-old female who had a left eye cataract extraction in another hospital, which provided us with a post-operation photo in the left eye and a pre-operation photo in the right eye (Fig. 2). According to her medical records, the patients have congenital posterior subcapsular opacities cataract with microcornea (Fig. 2). The axial length of her right eye is 24.70 mm and 24.03 mm in the left eye; the corneal diameter is 9.5 mm in both eyes (Table 1). In addition, the proband also has nystagmus and strabismus. Her mother, sister, and daughter also have similar phenotypes with the proband. All affected individuals were reported to be affected since birth. None of the family members had any other ocular or systemic abnormalities identified after a complete physical and ophthalmologic examination.

Pedigree of the congenital cataract with microcornea family. The affected family with autosomal dominant mode of inheritance. Open-square male; Open-circle female; Circle individuals with phenotype of congenital cataract with microcornea, arrowhead indicates the proband

Pre-operation eye photographs and pre-operation eye photographs of the proband with congenital cataract and microcornea in a Chinese family. A Pre-operation eye photographs of the proband. B Post-operation eye photographs of the proband. She had a left eye cataract extraction in another hospital, which provided us with post-operation photos
In silico analysis of variant in SOX2
After exome sequencing, data were filtered and analyzed for the causative variant of clinical phenotypes observed in patients. A missense variant, c.295G > T (p.A99S, NM_003106.4) in the SOX2 gene, was responsible for the disease. The SOX2 gene was located on chromosome 3 (3q26.33), encoding 317 amino acids. This variant has not been reported in genomAD, 1000 genomes databases or in the ExAC database. In predictive software, this missense variant was damaging in SIFT, MAPP, MutationTaster2, and PolyPhen-2, which showed the deleterious effect of the mutation on protein function (Table 2).
Segregation analysis
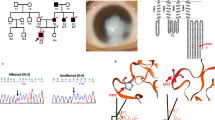
Sanger sequencing confirmed the autosomal dominant mode of inheritance of the SOX2 gene mutation (c.295G > T, p.A99S) in the affected family. All affected family members were heterozygous for this mutation, while all unaffected were homozygous normal (Fig. 3). Besides, this variant was absent in 200 unrelated controls, suggesting it is extremely rare.

DNA sequencing profiles of the identified mutations (upper) and their wild-type form (under)
In silico analysis
We carried out comparative and structural analyses to predict the potential pathogenic impact of p.Ala99Ser on protein level. This missense mutation causes the replacement of an Alanine with a Serine in the protein product of SOX2. The topological model revealed that SOX2 p.Ala99Ser was conserved in the high mobility group (HMG)-box domain (Fig. 4A). In addition, protein structure modeling showed that p.Ala99Ser was located in the helix and increased additional hydrogen bonds within space surrounded (Fig. 4B).

Mutations and predicted crystal structures of SOX2 protein. A Location of the mutations with respect to the topological model of the SOX2 polypeptide. B Predicted crystal structures of wild-type (left) and mutant (right) SOX2 protein. SOX family member contains a highly conserved HMG domain (in green/purple), with NLSs positioned within the extremities of the HMG domain
Discussion
Our study reports the identification of a novel missense mutation in SOX2, c.295G > T (p. Ala99Ser), in a Chinese family with congenital cataract and microcornea. More importantly, we reveal that missense mutation in SOX2 can cause a slight eye disorder, that is congenital cataract and microcornea, warranting further investigations on the pathogenesis of this disorder. This result expands the mutation spectrum of SOX2 and provides useful information to the study of the molecular pathogenesis of cataract and microcornea.
Normal dosage of the SOX2 gene are a developmentally regulated transcription factor that plays a critical role in embryonic development, particularly in the eyes and brain [17]. SOX2 is a single exon transcription factor previously associated with anophthalmia [18, 19], microphthalmia [20], and coloboma [21]. Sox2 is involved in crystallin regulation in murine [22] and avian models [23] and humans, and SOX2 mutations cause microphthalmia and cataracts [24, 25]. Previously, 21 of 37 patients with anophthalmia and microphthalmia carried frameshift and deletion/translocation mutations in the SOX2 gene [26]. In terms of missense mutations, only 2 of 37 patients with microphthalmia carried missense mutations, c.131C > G and c.166C > G [26, 27], indicating that phenotype released from deletion missense mutation. Interestingly, a missense mutation in this study, SOX2 c.295G > T p.Ala99Ser, caused a slight phenotype: microcornea and cataract.
Interestingly, this study reports that this novel mutation in SOX2 may lead to a broader phenotypic spectrum depending on the affected partner interaction domains. In addition to this, it is possible that the genetic makeup of these partner factors, other ocular development genes, or as yet unknown factors may play a role in the phenotypic expression of SOX2 mutations. Since expression during lens development represents an important site of SOX2 protein activity, some mutations may lead to phenotypes associated with specific loss of SOX2-related activity in the lens, such as cataracts and anterior segment defects. As mentioned above, the variation in phenotypes associated with, in particular, missense changes in SOX2, may be due to these mutations resulting in alteration of its interactions with tissue-specific protein partners rather than a complete loss-of-function [15]. The missense mutation in this study, SOX2 c.295G > T p.Ala99Ser, is conserved in the high mobility group (HMG)-box domain [15],which is critical for correct binding to interacting proteins and to target DNA sequences [28, 29]. The phenotype of associated microcornea may be due to the induction of corneal effects by an abnormally formed lens during embryonic development and/or a reduction of SOX2 on corneal molecular chaperones.
This study also has some limitations. Specifically, no functional analysis was performed of the identified variants in this study. Although gene function experiments and animal models are lacking, with the help of previous studies and using this as a basis, this deleterious mutation is pathogenic in combination with in silico analyses and structural comparison in SOX2 variants.
Conclusion
A novel missense mutation (c.295G > T, p.Ala99Ser) in the SOX2 gene was found in this Han Chinese family with congenital cataract and microcornea. Our study demonstrated the missense mutation in SOX2 was associated with CCMC, warranting further investigations on the pathogenesis of this disorder. This result expands the mutation spectrum of SOX2 and provides useful information to the study of the molecular pathogenesis of cataract and microcornea.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. And sequencing data have been submitted to NCBI SRA database: PRJNA797523.
References
Haargaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M. A nationwide Danish study of 1027 cases of congenital/infantile cataracts: etiological and clinical classifications. Ophthalmology. 2004;111(12):2292–8.
Sheeladevi S, Lawrenson JG, Fielder AR, Suttle CM. Global prevalence of childhood cataract: a systematic review. Eye (Lond). 2016;30(9):1160–9.
Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017;156:95–102.
Huang XF, **ang L, Cheng W, Cheng FF, He KW, Zhang BW, et al. Mutation of IPO13 causes recessive ocular coloboma, microphthalmia, and cataract. Exp Mol Med. 2018;50(4):1–11.
Lang E, Koller S, Bahr L, Toteberg-Harms M, Atac D, Roulez F, et al. Exome sequencing in a Swiss childhood Glaucoma cohort reveals CYP1B1 and FOXC1 variants as Most frequent causes. Transl Vis Sci Technol. 2020;9(7):47.
Willoughby CE, Shafiq A, Ferrini W, Chan LL, Billingsley G, Priston M, et al. CRYBB1 mutation associated with congenital cataract and microcornea. Mol Vis. 2005;11:587–93.
Hansen L, Yao W, Eiberg H, Kjaer KW, Baggesen K, Hejtmancik JF, et al. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci. 2007;48(9):3937–44.
Rechsteiner D, Issler L, Koller S, Lang E, Bahr L, Feil S, et al. Genetic analysis in a Swiss cohort of bilateral congenital cataract. JAMA Ophthalmol. 2021;139(7):691–700.
Shiels A, Bennett TM, Hejtmancik JF. Cat-map: putting cataract on the map. Mol Vis. 2010;16:2007–15.
Huang XF, Mao JY, Huang ZQ, Rao FQ, Cheng FF, Li FF, et al. Genome-wide detection of copy number variations in unsolved inherited retinal disease. Invest Ophthalmol Vis Sci. 2017;58(1):424–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS, et al. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29(11):1265–72.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–85.
Jagga B, Edwards M, Pagin M, Wagstaff KM, Aragao D, Roman N, et al. Structural basis for nuclear import selectivity of pioneer transcription factor SOX2. Nat Commun. 2021;12(1):28.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, et al. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33(4):461–3.
Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, et al. SOX2 anophthalmia syndrome. Am J Med Genet A. 2005;135(1):1–7 discussion 8.
Schneider A, Bardakjian T, Reis LM, Tyler RC, Semina EV. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet A. 2009;149A(12):2706–15.
Wang P, Liang X, Yi J, Zhang Q. Novel SOX2 mutation associated with ocular coloboma in a Chinese family. Arch Ophthalmol. 2008;126(5):709–13.
Cvekl A, McGreal R, Liu W. Lens development and Crystallin gene expression. Prog Mol Biol Transl Sci. 2015;134:129–67.
Shimada N, Aya-Murata T, Reza HM, Yasuda K. Cooperative action between L-Maf and Sox2 on delta-crystallin gene expression during chick lens development. Mech Dev. 2003;120(4):455–65.
Donner AL, Episkopou V, Maas RL. Sox2 and Pou2f1 interact to control lens and olfactory placode development. Dev Biol. 2007;303(2):784–99.
Kondoh H, Uchikawa M, Kamachi Y. Interplay of Pax6 and SOX2 in lens development as a paradigm of genetic switch mechanisms for cell differentiation. Int J Dev Biol. 2004;48(8-9):819–27.
Amlie-Wolf L, Bardakjian T, Kopinsky SM, Reis LM, Semina EV, Schneider A. Review of 37 patients with SOX2 pathogenic variants collected by the Anophthalmia/Microphthalmia clinical registry and DNA research study. Am J Med Genet A. 2022;188(1):187–98.
Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, et al. Familial recurrence of SOX2 anophthalmia syndrome: phenotypically normal mother with two affected daughters. Am J Med Genet A. 2008;146A(21):2794–8.
Kamachi Y, Uchikawa M, Kondoh H. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. 2000;16(4):182–7.
Wilson M, Koopman P. Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev. 2002;12(4):441–6.
Acknowledgements
The authors thank the affected patients and their parents for their participation in the study. The authors acknowledge the entire staff of the Eye hospital for their support.
Funding
**an municipal science and technology project (No. 2019114613YX001SF034(2) to LY); Key research and development program of Shaanxi province (No. 2018SF-066 to LY).
Author information
Authors and Affiliations
Contributions
ZBL, JL, and FFL contributed to the study design. ZBL and HSS contributed to sample and clinical data collection. ZBL, JL, HSS, AYY, SHC, and FFL contributed to data interpretation. ZBL, FFL, LY wrote the main manuscript text. All authors reviewed the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethical Committee of the Eye Hospital of Wenzhou Medical University and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from parents of the affected individuals for blood sample collection.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, ZB., Li, J., Ye, L. et al. Novel SOX2 mutation in autosomal dominant cataract-microcornea syndrome. BMC Ophthalmol 22, 70 (2022). https://doi.org/10.1186/s12886-022-02291-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02291-4