
Abstract
Background
Epilepsy, a challenging neurological condition, is often present with comorbidities that significantly impact diagnosis and management. In the Pakistani population, where financial limitations and geographical challenges hinder access to advanced diagnostic methods, understanding the genetic underpinnings of epilepsy and its associated conditions becomes crucial.
Methods
This study investigated four distinct Pakistani families, each presenting with epilepsy and a spectrum of comorbidities, using a combination of whole exome sequencing (WES) and Sanger sequencing. The epileptic patients were prescribed multiple antiseizure medications (ASMs), yet their seizures persist, indicating the challenging nature of ASM-resistant epilepsy.
Results
Identified genetic variants contributed to a diverse range of clinical phenotypes. In the family 1, which presented with epilepsy, developmental delay (DD), sleep disturbance, and aggressive behavior, a homozygous splice site variant, c.1339–6 C > T, in the COL18A1 gene was detected. The family 2 exhibited epilepsy, intellectual disability (ID), DD, and anxiety phenotypes, a homozygous missense variant, c.344T > A (p. Val115Glu), in the UFSP2 gene was identified. In family 3, which displayed epilepsy, ataxia, ID, DD, and speech impediment, a novel homozygous frameshift variant, c.1926_1941del (p. Tyr643MetfsX2), in the ZFYVE26 gene was found. Lastly, family 4 was presented with epilepsy, ID, DD, deafness, drooling, speech impediment, hypotonia, and a weak cry. A homozygous missense variant, c.1208 C > A (p. Ala403Glu), in the ATP13A2 gene was identified.
Conclusion
This study highlights the genetic heterogeneity in ASM-resistant epilepsy and comorbidities among Pakistani families, emphasizing the importance of genotype-phenotype correlation and the necessity for expanded genetic testing in complex clinical cases.
Similar content being viewed by others

Background
Epilepsy is a neurological disease characterized by the presence of recurrent and unprovoked seizures, reflecting underlying brain dysfunction. While seizures are the hallmark of epilepsy, the clinical landscape is often further complicated by the presence of comorbidities—additional medical, cognitive, or behavioral conditions that frequently accompany epilepsy [1, 2]. These comorbidities introduce complexity to the clinical landscape, making it challenging to understand and manage the overall situation. They often lead to more severe phenotypes in epilepsy patients and are associated with negative outcomes, including a poor prognosis, inadequate responses to ASMs, reduced quality of life, and increased mortality [3]. The intricate relationship between epilepsy and its associated comorbidities presents a formidable challenge in neurological research, especially in populations with limited access to advanced diagnostic techniques. In such settings, understanding the genetic components of epilepsy and its associated comorbidities is crucial for effective diagnosis and management. This challenge is particularly relevant in the context of the Pakistani population, characterized by financial limitations and geographical obstacles. Despite the lack of readily available genetic testing services in Pakistan, and the substantial costs associated with high-throughput approaches such as WES, investigating the genetic architecture of epilepsy and associated comorbidities remains imperative. It is essential for a comprehensive understanding of disease pathophysiology and the development of possible precision medicine-based approaches for better clinical management.
Importantly, the occurrence of comorbidities in epileptic patients reflects an overlap of causative genes and the involvement of similar molecular pathways [4, 5]. Previous research has provided convincing evidence that specific genetic variations may play a role in both epilepsy and its associated conditions within individuals. For instance, variants in genes such as NRXN1, SCN1A, PTEN, TBCD, ARFGEF1, GNAI1, SYNGAP1, GRIN2A, SPTBN5, and CNTNAP2 have been linked to epilepsy as well as neurobehavioral and neurodevelopmental comorbidities or other related symptoms [1, 6,7,8,9,10,11,12,13,14,15]. The range of comorbidities linked to epilepsy is diverse; a single gene variant may lead to outcomes such as developmental delay (DD), intellectual disability (ID), and various other manifestations, each with differing severity. Additionally, it has been noted that within the same family, individuals carrying identical genetic variants may exhibit a spectrum of diverse phenotypes. For instance, one family member may experience severe, treatment-resistant seizures indicative of ASM-resistant epilepsy, while another may present with milder forms of epileptic seizures [6]. This highlights the complex nature of ASM-resistant epilepsy and its associated conditions, which can be influenced by genetic factors, impacting various aspects of an individual’s health and development.
Therefore, studies investigating familial occurrences of epilepsy may reveal genotypic and phenotypic heterogeneity of epilepsy as well as its comorbidities. Few studies have found an association of genetic variants with epilepsy and its comorbid conditions in families of Pakistani origin. In one study, a missense CNTNAP2 variant was identified in two consanguineous Pakistani families with epilepsy, DD, ID, and aggressive behavior. Similarly, in another study, a missense SPTBN5 variant was detected in a Pakistani family with early-onset epilepsy, behavioral impairments, and gastroesophageal reflux [7]. Recently, homozygous variants in OCLN, ALDH7A1, IQSEC2, COL3A1, CNTNAP2, TRIT1, and NARS1 were identified in individuals from Pakistani families who presented with epilepsy and frequently accompanied DD phenotype [8]. These studies have unveiled a notable lack of genetic investigations specifically targeting families of Pakistani origin from the Khyber Pakhtunkhwa region. This highlights the urgent need to prioritize this geographical area in future research efforts to comprehensively understand the genetic aspects of epilepsy and its management within this population subset. Therefore, in current study, we focused primarily on investigating the genetic causes of epilepsy and its associated comorbidities within four Pakistani families from the Khyber Pakhtunkhwa region. Our investigation revealed diverse clinical phenotypes and distinct homozygous genetic variants associated with specific sets of symptoms, emphasizing the challenging nature of ASM-resistant epilepsy among family members residing in this region.
Methods
Study population
The current study was approved by the Ethical Committee as well as the Advance Studies and Research Board of Kohat University of Science and Technology. Families residing in Khyber Pakhtunkhwa, Pakistan, with at least two affected members were eligible for inclusion in the study. The diagnosis of epilepsy was made by Pakistan Medical and Dental Council (PMDC) recognized and qualified epileptologists/neurologists at Khyber Teaching Hospital in Peshawar. It relied primarily on clinical features, including history and examination, with electroencephalography (EEG) as the diagnostic test. Patients resistant to successive lines of drug treatment were classified as ASM-resistant in accordance with the criteria established by the International League Against Epilepsy (ILAE) [9], with consideration given to related variations where indicated. After obtaining written informed consent for genetic analysis, blood samples were collected from all the affected and unaffected family members included.
Genetic studies
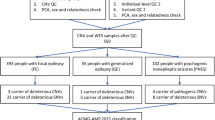
Whole Exome Sequencing (WES) was performed on Genomic DNA of affected individuals (III:4 and III:6 in family1, IV:6 in family2, III:1 in family3 and III:2 in family4). Libraries for each individual were obtained using the xGen DNA Library Prep Kit EZ. Subsequently, they were enriched using the xGen™ Exome Hyb Panel v2 and xGen™ Hybridization and Wash Kit, following the manufacturer’s instructions. Enriched libraries were finally sequenced on the Illumina Novaseq platform, using 151 bp paired-end reads. Sequence data in the form of BAM files were generated via the Picard data-processing pipeline and contained well-calibrated reads aligned to the GRCh37 human Genome reference. Additionally, quality control (QC) statistics for the WES run were assessed (Supplementary Data File 1: Table). Samples across projects were then jointly called via the Genome Analysis Toolkit (GATK) best-practice pipeline31 for data harmonization and variant discovery. This pipeline detected single-nucleotide variants (SNVs) and small insertion or deletion (indel) variants from exome sequence data [10]. All the variants were screened according to the location, frequency, and type of variation. Variants were filtered with a minor allele frequency (MAF) cutoff of < 0.01% in the Exome Variant Server (http://evs.gs.washington.edu/EVS/), GnomAD (https://gnmad.broadinstitute.org), and 1000 Genomes (http://www.1000genmes.org/).
Alleles specific primers (Supplementary Data File 2: Table) were designed using primer3 software (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) for the validation and segregation analysis, which involved polymerase chain reaction (PCR) and Sanger sequencing. The amplified PCR products were sequenced by Bei**g Tsingke Biotech Co., Ltd. (https://tsingke.com/pages/about-us-1). The Sanger sequencing analysis was carried out using Chromas Lite version 2.6.6, a chromatogram viewer software. The pathogenicity of segregating genetic variants was predicted using various in silico tools: PredictSnp (https://loschmidt.chemi.muni.cz/predictsnp/), Sift (https://sift.bii.a-star.edu.sg/), Mutation assessor (http://mutationassessor.org/r3/) Mutation taster (https://www.mutationtaster.org/), SpliceAI (https://spliceailookup.broadinstitute.org/) and MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) SPiP (https://sourceforge.net/projects/splicing-prediction-pipeline/). PhyloP scores for measuring evolutionary conservation were obtained from the UCSC Genome Browser (https://genome.ucsc.edu/). Genetic variants were classified and interpreted using the American College of Medical Genetics and Genomics (ACMG) standards and guidelines [11]. In order to provide a clearer insight into the impact of the variants on the patients’ clinical condition, all reported genetic variants were retrieved from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) HGMD (http://www.hgmd.cf.ac.uk/ac/search.php), PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), and OMIM (https://www.ncbi.nlm.nih.gov/omim/) databases.
Results
In current study, we observed a significant genetic heterogeneity in four Pakistani families with ASM- resistant epilepsy and associated comorbidities, revealing noteworthy clinical phenotype variations both within and between families. The comorbid conditions identified in affected individuals from four epilepsy families are displayed in Table 1. It was determined that these comorbidities had an underlying genetic etiology and were associated with autosomal recessive inheritance of COL18A1, ZFYVE26, UFSP2, and ATP13A2 variants. Table 2 displays the localization, frequency (gnomAD), predictions, PhyloP scores, pathogenicity (ACMG class), and novelty status of segregating genetic variants. These variants were found within long-sized regions of homozygosity (Supplementary Data File 3: Tables) and demonstrated associations with distinct sets of symptoms and clinical phenotypes.
Figure 1 displays the number of ASMs used in history by the epileptic patients in the investigated families. It’s noteworthy that these patients were prescribed multiple ASMs in their treatment efforts, yet their seizures persisted, indicating cases of ASM-resistant epilepsy. Similarly, Fig. 2 depicts the frequency of ASMs usage by epileptic patients in investigated families, and notably, Valproate Sodium was the most used ASM by Pakistani epileptic patients.

The number of ASMs used in history by the epileptic patients in investigated families

The frequency of ASMs usage by epileptic patients in investigated families
Family 1
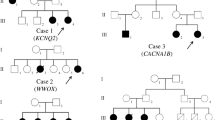
The proband of family-1 was a 20-year-old male (III:4) with epilepsy, DD, insomnia, and aggressive behavior born from non-consanguineous parents. He experienced the first unprovoked seizure at the age of 5 years. His seizures control could not be achieved with prescribed ASMs. The interictal EEG shows generalized myoclonic seizures. The affected siblings (III:2, III:3 & III:4) and cousins (III:6, III:7, III:8 & III:9) were exhibiting similar disease features. DD during childhood was a prevalent feature among them. Furthermore, as individuals aged, they exhibited symptoms like insomnia and increased aggression (Table 1).
WES analysis revealed a novel homozygous splice site variant [Chr21 (GRCh37): g.46,896,259 C > T NM_001379500.1: c.1339–6 C > T] in COL18A1 in the proband. In silico analysis predicted it as ‘likely benign,’ as the PhyloP score indicated a low level of evolutionary conservation at this position. However, Sanger sequencing verified the segregation of the COL18A1 variant with the observed phenotype, confirming its homozygous status in the proband, his siblings, and cousins (Fig. 3.1a & b). This variant consistently manifested as a recessive allele in affected family members and was never observed in a homozygous state within the control population database. The rarity of this variant in its monoallelic status in GnomAD (MAF = 0.0000323) emphasizes its potential as a cause of an autosomal recessive disorder (Table 2). Based on these factors, the COL18A1 c.1339–6 C > T variant is classified as a Variant of Uncertain Significance (VUS) in accordance with ACMG guidelines (Table 2).

Families pedigrees and genetic findings: 1a. Pedigree of family 1, 1b. showing segregation of the identified variant in COL18A1 gene, 1c. Homozygosity map of the proband (III:6), showing the runs of homozygosity as blue bands. 2a. Pedigree of family 2, 2b. showing segregation of the identified variant in UFSP2 gene, 2c. Homozygosity map of the proband (IV:5), showing the runs of homozygosity as blue bands. 3a. Pedigree of family 3, 3b. showing segregation of the identified variant in ZFYVE26 gene. 4a. Pedigree of family 4, 4b. showing segregation of the identified variant in ATP13A2 gene, 4c. Homozygosity map of the proband (III:2), showing the runs of homozygosity as blue bands
Family 2
The proband (IV:5) in family-2, a 21-year-old male was born from consanguineous healthy parents (Fig. 3.2a). The proband experienced the first unprovoked seizure at the age of 14 years. In addition, he did not respond to ASM. Interictal EEG shows generalized clonic tonic seizure. Brain computed tomography scan was normal (Table 1). Epilepsy-associated comorbidities observed in the proband included DD, ID, and anxiety. His 25-year-old brother has similar findings (Table 1). His sister (IV:1) and aunt (II:1) both deceased due to status epilepticus during childhood.
By WES analysis, a homozygous missense variant [Chr4(GRCh37): g.186,337,011 A > T, NM_018359.5: c.344T > A, p. Val115Glu] in UFSP2 gene was identified in the proband. The segregation study verified the inheritance pattern of the UFSP2 variant, confirming its homozygous status in both the proband and his brother (Fig. 3.2a & b). The variant c.344T > A in UFSP2 gene with frequency (MAF = 0.0000713) was listed in the gnomAD that leads to a substitution of Valine with Glutamic Acid and at the evolutionary conserved position 115 (p. Val115Glu) according to the UCSC Human Genome (GRCh37/ hg19) and Ensemble database. In addition, this variant is predicted as pathogenic by ACMG and likely pathogenic by in silico analysis (Table 2). The variant has already been described in the literature as pathogenic and associated with pediatric neurodevelopmental anomalies and epilepsy phenotype (PMID: 33,473,208).
Family 3
In Family-3, a proband, a 7-year-old child (III:1), who presented a complex clinical phenotype. The proband’s medical history, as revealed through the interictal EEG examinations, indicated the onset of generalized tonic-clonic seizures at an exceptionally early age of one month. Additionally, the proband exhibited symptoms of epilepsy, ataxia, ID, and DD (Table 1). Notably, both parents were phenotypically normal, despite their consanguineous relationship. The grandfather (I:1) of the proband had similar features of epilepsy and ataxia but did not exhibit ID and DD (Fig. 3.3a).
WES analysis was conducted, revealing a novel homozygous frameshift variant, [Chr14 (GRCh37): g.68265038GGCTTGGCATTGTATAA > G NM_015346.4: c.1926_1941del], in the ZFYVE26 gene within the proband. The validation studies verified its homozygous status in both the proband and his grandfather (Fig. 3.3a & b). Based on ACMG criteria and in silico analysis, the variant, denoted as c.1926_1941del, is predicted to be a “variant of likely pathogenic”. Remarkably, this variant consistently presented as a recessive allele in all affected family members and was not detected in a homozygous state within control population databases (Table 2). Its absence in its monoallelic status in both the Genome Aggregation Database (GnomAD) and the 1000 Genomes Project further underscores its potential pathogenic role in the context of an autosomal recessive disorder. These findings suggest a compelling link between the ZFYVE26 variant and the observed clinical phenotypes in the proband and his grandfather, shedding light on its likely pathogenic impact.
Family 4
Within the scope of this study, we investigated family 4, which presented a remarkable clinical complexity. The proband (III:2), a nearly 10-year-old child, displayed a constellation of symptoms, including epilepsy, Parkinson’s disease (PD), ID, DD, deafness, excessive drooling, speech impediment, hypotonia, and a notably weak cry (Table 1). Seizures first manifested at a remarkably early age of just one month, with interictal EEG reports indicating generalized myoclonal seizures. Intriguingly, the parents of the proband had a consanguineous relationship (Fig. 3.4a), yet both showed no overt phenotypic abnormalities. Within the same family, an affected sibling (III:1) displayed similar clinical characteristics, including epilepsy, Parkinson’s disease, speech impediment, and deafness, but did not experience ID or DD (Table 1).
WES analysis was systematically performed, leading to the identification of a homozygous missense variant, [Chr1 (GRCh37): g.17,322,979 C > A NM_022089.4: c.1208 C > A_p.Ala403Glu], localized within the ATP13A2 gene, in the proband of the studied family. To validate the inheritance pattern of this ATP13A2 variant and its connection with the manifested clinical phenotypes, Sanger sequencing was conducted, conclusively affirming its homozygous status in both the proband and a sibling (Fig. 3.4a & b). Based on ACMG criteria and in silico analysis, the variant c.1208 C > A, is classified as a VUS and ‘damaging’ respectively (Table 2). Remarkably, this particular genetic alteration consistently demonstrated a recessive mode of inheritance across all afflicted family members and remained conspicuously absent in the homozygous state within population databases utilized for control comparisons. Its rare occurrence in the heterozygous state, as evident in both the GnomAD (MAF = 0.0000297) databases, further underscores its potential implication as a pathogenic contributor within the framework of an autosomal recessive disorder. These findings collectively offer compelling evidence of an association between the ATP13A2 variant, and the clinical features observed in the proband, thus highlighting its plausible pathogenic role.
Discussion
Investigation of epilepsy and comorbidities in the Pakistani population has the potential to contribute not only to the advancement of personalized medical approaches but also to the enrichment of our knowledge about the underlying genetic factors and clinical profiles associated with this challenging neurological condition. Typically, family history, clinical assessments, and investigations using EEG/video-EEG telemetry and brain imaging (such as computed tomography or magnetic resonance imaging) can establish a clinical diagnosis in epilepsy patients. Nevertheless, many of these clinical diagnostic approaches are beyond the reach of patients in remote regions of Pakistan due to financial limitations and a lack of availability at their residential areas. Moreover, the presence and variable phenotypes of comorbidities in patients with epilepsy pose significant challenges for diagnosis and management [12]. Importantly, the use of next-generation sequencing (NGS) technologies, such as WES, is extremely useful in determining the underlying genetic causes of epilepsy and its comorbid conditions in families. Using WES, homozygous pathogenic variants in the COL18A1, UFSP2, ZFYVE26 and ATP13A2 genes were detected to cause ASM-resistant epilepsy and its comorbid conditions in four Pakistani families. While acknowledging the limitations and costs associated with genetic testing, particularly in resource-limited settings, our findings emphasize the importance of genetic investigations in elucidating the molecular basis of ASM-resistant epilepsy and guiding precision medicine-based approaches for improved clinical management.
The COL18A1 gene encodes the α1 chain of collagen type XVIII, which is a component of basement membranes, specialized structures critical for the normal development of various tissues [13]. Different tissues have specific requirements for collagen composition and organization [14]. Importantly, Collagen XVIII is necessary for the normal development of the brain and eye. Consequently, disrupted function of this protein can result in central nervous system (CNS) and eye defects, respectively [13]. Biallelic variants in the COL18A1 gene were first described to cause a very rare autosomal recessive syndrome known as Knobloch syndrome (KS), characterized by a range of ocular, genitourinary and CNS defects [14]. Since the first report, many studies have associated COL18A1 variants with KS (Supplementary Data File 4: Figure) and have observed variations in phenotype among patients with KS [15,26]. These findings of present and previous studies highlight the intricate interplay between genetic variations in COL18A1 and the development of epilepsy and associated conditions.
Individuals may experience different combinations of disease features depending on the effects of specific COL18A1 variants on the protein’s structure and function. Among the reported pathogenic COL18A1 variants in homozygous and compound heterozygous states in KS families in previous studies, the majority of them were described as loss-of-function variants causing protein truncation, leading to KS [15, 18, 27]. Previously, compound heterozygous COL18A1 variants were identified in patients with CNS defects such as autism and developmental delay without ocular malformations. Expression analysis in these studies revealed that residual expression of COL18A1 in eye cells allows normal ocular development. Thus, it was inferred that compound heterozygous COL18A1 variants do not completely abolish COL18A1 expression [21, 30] and also releases UFM1 from UFMylated proteins in a process known as de-UFMylation [31, 32]. Both UFMylation and de-UFMylation are processes of post-translational protein modification, and functional studies have revealed that the loss of key components of these processes results in defects in embryogenesis, hematopoiesis, cellular differentiation, and brain development [30, 41,42]. In some cases, it may also be accompanied by distal amyotrophy, pigmentary maculopathy, and atypical parkinsonism [43, 44]. Here, we expand the phenotypic spectrum associated with ZFYVE26 variants, by reporting a Pakistani epileptic family with spastic ataxia DD and ID phenotypes. Most of the variants identified in ZFYVE26 are frameshift or nonsense variants, indicating a loss-of-function mechanism in the pathogenesis of AR HSP [42]. Similarly, the ZFYVE26 variant p.Tyr643MetfsX2 identified in the current study is a frameshift variant resulting in a truncated protein, disrupting the normal function of the gene and, consequently, causing ASM-resistant epilepsy, spastic ataxia, DD, and ID phenotypes in patients. The findings of both the previous and current studies expand our understanding of the phenotypic spectrum associated with ZFYVE26 variants and affirm the idea that the ZFYVE26 variants primarily result in a loss-of-function mechanism in the development of neurodegenerative and neurodevelopmental phenotypes.
The ATP13A2 gene encodes a lysosome-associated transmembrane enzyme that restores lysosomal function under physiological conditions. This enzyme shows high expression in the human brain [45] and loss-of-function ATP13A2 variants have been reported to cause rare autosomal recessive juvenile-onset neurodevelopmental and neurodegenerative conditions. These conditions include Kufor-Rakeb syndrome [46], spastic paraplegia [47], neuronal ceroid lipofuscinoses [48], and amyotrophic lateral sclerosis [49]. In patients with these phenotypes, lysosomal function is impaired due to missense or nonsense ATP13A2 variants. Recent research has revealed that missense ATP13A2 variants cause misfolding of the ATP13A2 protein. The misfolding of the ATP13A2 protein relocates it from its original lysosomal position to the endoplasmic reticulum (ER) [50,51,52]. The ER serves a crucial role in eliminating unwanted and potentially harmful proteins [53]. Once the misfolded ATP13A2 protein is in the ER, it triggers the endoplasmic reticulum-associated degradation (ERAD) pathway, resulting in its rapid degradation. This process ultimately leads to a decrease in ATP13A2 protein levels within lysosomes [54, 55]. In the case of a nonsense ATP13A2 variant, the transcribed mRNA triggers the nonsense-mediated mRNA decay (NMD) pathway due to the presence of a premature termination codon. Eventually, the nonsense ATP13A2 mRNA is degraded, also leading to a decrease in ATP13A2 protein levels within lysosomes [51, 56]. The ERAD and NMD pathways serve as the molecular basis for the loss-of-function nature of ATP13A2 variants.
In the current study, we identified a homozygous missense variant in the ATP13A2 gene, specifically c.1208 C > A, p.Ala403Glu, in a family displaying a range of symptoms, including ASM-resistant epilepsy, ID, DD, PD, deafness, drooling, speech impediments, hypotonia, and a weak cry. This particular ATP13A2 variant had previously been classified as a VUS within the context of juvenile-onset PD syndrome (Kufor-Rakeb syndrome) in the ClinVar Miner database (ClinVar; SCV000351396). However, our segregation analysis in a Pakistani family has, for the first time, established the pathogenic nature of this ATP13A2 variant in relation to the observed disease phenotypes. The variant was consistently identified as a recessive allele in affected individuals and was absent in unaffected individuals. Furthermore, it exhibited an exceedingly low frequency in gnomAD (MAF = 0.0000297), providing compelling evidence of its pathogenicity. Consequently, our study strongly supports the hypothesis that the ATP13A2 variant is causative for ASM-resistant epilepsy and the associated comorbidities, including PD.
In conclusion, this study highlights the genetic and clinical heterogeneity associated with ASM-resistant epilepsy and its comorbidities in the Pakistani population. Pathogenic variants in COL18A1, UFSP2, ZFYVE26, and ATP13A2 contribute to a wide range of clinical phenotypes, from ASM-resistant epilepsy and developmental delay to ataxia, intellectual disability, speech impediment, and other associated symptoms. The findings underscore the importance of genotype-phenotype correlation and the need for expanded genetic testing, especially in challenging clinical cases. Addressing financial and geographical barriers to healthcare access in remote regions of Pakistan is essential for improved patient care. Moreover, further research into the functional consequences of specific genetic variants and the establishment of rare disease registries can advance our understanding and support for individuals with epilepsy and comorbid conditions.
Data availability
The datasets generated and/or analyzed during the current study are available in the ClinVar repository, with the accession numbers i.e., SCV004174822, SCV004174821, SCV004174820, and SCV004174819.
References
Shimizu H, Morimoto Y, Yamamoto N, Tayama T, Ozawa H, Imamura AJEP. Overlap between epilepsy and neurodevelopmental disorders: insights from clinical and genetic studies. 2022:41–54.
Sorg A-L, von Kries R, Borggraefe IJJN. Cognitive disorders in childhood epilepsy: a comparative longitudinal study using administrative healthcare data. 2022;269(7):3789–99.
Møller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. 2016;7(4):210–9.
O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome Sequencing Sporadic Autism Spectr Disorders Identifies Severe de novo Mutations. 2011;43(6):585–9.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong Association de novo copy Number Mutations Autism. 2007;316(5823):445–9.
Ellis CA, Petrovski S, Berkovic SFJTLN. Epilepsy genetics: clinical impacts and biological insights. 2020;19(1):93–100.
Khan A, Bruno LP, Alomar F, Umair M, Pinto AM, Khan AA et al. SPTBN5, encoding the βV-spectrin protein, leads to a syndrome of intellectual disability, developmental delay, and seizures. 2022;15:877258.
Shafique A, Sultan T, Alzahrani F, Seo GH, Alkuraya FS, Naz SJG. Genomic analysis of multiplex consanguineous families reveals causes of neurodevelopmental disorders with epilepsy. 2023;879:147599.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on therapeutic strategies. Wiley Online Library; 2010.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. 2013;43(1):110. 1-.0. 33.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 2015;17(5):405–23.
Mula MJEron. The comorbidities of epilepsy explained. 2020;20(12):1207–9.
Heljasvaara R, Aikio M, Ruotsalainen H, Pihlajaniemi TJMB. Collagen XVIII in tissue homeostasis and dysregulation—lessons learned from model organisms and human patients. 2017;57:55–75.
Suzuki OT, Sertié AL, Der Kaloustian VM, Kok F, Carpenter M, Murray J et al. Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. 2002;71(6):1320–9.
Li S, Wang Y, Sun L, Yan W, Huang L, Zhang Z et al. Knobloch Syndrome Associated with Novel COL18A1 variants in Chinese Population. 2021;12(10):1512.
Wang P, Jia X, **ao X, Li S, Long Y, Liu M et al. An early diagnostic clue for COL18A1-and LAMA1-associated diseases: high myopia with Alopecia Areata in the cranial midline. 2021;9:644947.
Levinger N, Hendler K, Banin E, Hanany M, Kimchi A, Mechoulam H et al. Variable phenotype of Knobloch syndrome due to biallelic COL18A1 mutations in children. 2021;31(6):3349–54.
Hull S, Arno G, Ku CA, Ge Z, Waseem N, Chandra A et al. Molecular and clinical findings in patients with Knobloch syndrome. 2016;134(7):753–62.
Caglayan AO, Baranoski JF, Aktar F, Han W, Tuysuz B, Guzel A et al. Brain malformations associated with Knobloch syndrome—review of literature, expanding clinical spectrum, and identification of novel mutations. 2014;51(6):806–13. e8.
Balikova I, Sanak NS, Fanny D, Smits G, Soblet J, De Baere E, et al. Three Cases Molecularly Confirmed Knobloch Syndrome. 2020;41(1):83–7.
Irene Díez García-Prieto I, Lopez-Martín S, Albert J, Jiménez de la Peña M, Fernández-Mayoralas DM, Calleja-Pérez B et al. Mutations in the COL18A1 gen associated with knobloch syndrome and structural brain anomalies: a novel case report and literature review of neuroimaging findings. 2022;28(1):11–8.
Corbett MA, Turner SJ, Gardner A, Silver J, Stankovich J, Leventer RJ et al. Familial epilepsy with anterior polymicrogyria as a presentation of COL18A1 mutations. 2017;60(8):437–43.
Weatherburn CJ, Heath CA, Mercer SW, Guthrie BJS. Physical and mental health comorbidities of epilepsy: population-based cross-sectional analysis of 1.5 million people in Scotland. 2017;45:125–31.
Villanueva-Mendoza C, Tuson M, Apam-Garduño D, de Castro-Miró M, Tonda R, Trotta JR et al. The genetic landscape of inherited retinal diseases in a Mexican cohort: genes, mutations and phenotypes. 2021;12(11):1824.
Friedman D, Kannan K, Faustin A, Shroff S, Thomas C, Heguy A et al. Cardiac arrhythmia and neuroexcitability gene variants in resected brain tissue from patients with sudden unexpected death in epilepsy (SUDEP). 2018;3(1):9.
Paisán-Ruiz C, Scopes G, Lee P, Houlden H. Homozygosity map** through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. 2009; 150B(7): 993–7.
Suzuki O, Kague E, Bagatini K, Tu H, Heljasvaara R, Carvalhaes L, et al. Novel Pathogenic Mutations skin Biopsy Anal Knobloch Syndrome. 2009;15:801.
Duan Y, Lin S, **e L, Zheng K, Chen S, Song H et al. Exome sequencing identifies a novel mutation of the GDI1 gene in a Chinese non-syndromic X-linked intellectual disability family. 2017;40:591–6.
Karbassi I, Maston GA, Love A, DiVincenzo C, Braastad CD, Elzinga CD et al. A standardized DNA variant scoring system for pathogenicity assessments in mendelian disorders. 2016;37(1):127–34.
Gerakis Y, Quintero M, Li H, Hetz CJT. UFMylation Syst Proteostasis beyond. 2019;29(12):974–86.
Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. 2007;282(8):5256–62.
Ishimura R, Obata M, Kageyama S, Daniel J, Tanaka K, Komatsu MJF. A novel approach to assess the ubiquitin-fold modifier 1‐system in cells. 2017;591(1):196–204.
Ni M, Afroze B, **ng C, Pan C, Shao Y, Cai L et al. A pathogenic UFSP2 variant in an autosomal recessive form of pediatric neurodevelopmental anomalies and epilepsy. 2021;23(5):900–8.
Di Rocco M, Rusmini M, Caroli F, Madeo A, Bertamino M, Marre-Brunenghi G et al. Novel spondyloepimetaphyseal dysplasia due to UFSP2 gene mutation. 2018;93(3):671–4.
Zhang G, Tang S, Wang H, Pan H, Zhang W, Huang Y et al. UFSP2-related spondyloepimetaphyseal dysplasia: a confirmatory report. 2020;63(11):104021.
Mattern L, Begemann M, Delbrück H, Holschbach P, Schröder S, Schacht SM, et al. Variant of the catalytic cysteine of UFSP2 leads to spondyloepimetaphyseal dysplasia type. Di Rocco. 2023;18:101683.
Raha S, Kotecha U, Mistri M, Shah P, Sharda SJMR. Familial infantile spasm syndrome due to biallelic variants in the gene encoding UFM1-specific peptidase 2 (UFSP2). 2023:100002.
Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. 2013;136(10):3119–39.
Vantaggiato C, Panzeri E, Castelli M, Citterio A, Arnoldi A, Santorelli FM et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. 2019;15(1):34–57.
Goizet C, Boukhris A, Maltete D, Guyant-Maréchal L, Truchetto J, Mundwiller E et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. 2009;73(14):1111–9.
Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L et al. Overlap** phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. 2014;137(7):1907–20.
Hsu S-L, Lu Y-J, Tsai Y-S, Chao H-C, Fuh J-L, Liao Y-C et al. Investigating ZFYVE26 mutations in a Taiwanese cohort with hereditary spastic paraplegia. 2022;121(1):126–33.
Mallaret M, Lagha-Boukbiza O, Biskup S, Namer IJ, Rudolf G, Anheim M et al. SPG15: a cause of juvenile atypical levodopa responsive parkinsonism. 2014;261:435–7.
Özdemir TR, Gençpınar P, Arıcan P, Öztekin Ö, Dündar NO, Özyılmaz BJIJN. A case of spastic paraplegia-15 with a novel pathogenic variant in ZFYVE26 gene. 2019;129(12):1198–202.
Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E et al. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. 2012;21(8):1725–43.
Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. 2010;25(12):1929–37.
Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). 2017;140(2):287–305.
Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJJH. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. 2012;21(12):2646–50.
Spataro R, Kousi M, Farhan SM, Willer JR, Ross JP, Dion PA et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. 2019;13(1):1–10.
Park JS, Blair NF, Sue CMJMD. The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. 2015;30(6):770–9.
Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor‐Rakeb syndrome, a form of early‐onset parkinsonism. 2011;32(8):956–64.
Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. 2011;286(34):29654–62.
Wan S-X, Pan X, Qian J-J, Shu Y, Xu P, Zhao J et al. Downregulation of ATP13A2 in midbrain dopaminergic neurons is related to defective autophagy in a mouse model of Parkinson’s disease. 2020;13(7):1853.
Covy JP, Waxman EA, Giasson BIJJ. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. 2012;90(12):2306–16.
Matsui H, Sato F, Sato S, Koike M, Taruno Y, Saiki S et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. 2013;587(9):1316–25.
Huang J, Xu S, Yu Z, Zheng Y, Yang B, Ou QJJGS. ATP13A2 is a prognostic biomarker and correlates with Immune infiltrates in Hepatocellular Carcinoma. 2023;27(1):56–66.
Acknowledgements
The authors would like to thank the patients and their family members for participation in the study.
Funding
This study received support from the Kohat University of Science and Technology in Kohat, Pakistan, as well as from the Higher Education Commission (HEC), Pakistan, through the International Research Support Initiative Program (IRSIP) fellowship awarded to Muhammad Yasin (1–8/HEC/HRD/2022/8802/PIN: IRSIP 50 BMS 40) and IRCCS, Istituto delle Scienze Neurologiche of Bologna; Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy.
Author information
Authors and Affiliations
Contributions
Clinical data collection, collation, and analysis: MY, IU, ZJ, LL and SS; Genetic testing and data analysis: MY, MD, ANA, AA, RM, LL, NK, VC and SS; Manuscript writing: MY, MD, NK and SS; Manuscript revision; LL, RM and VC; Study supervision and coordination: VC, NK and SS. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
The study was approved by the Ethical Committee of Kohat University of Science and Technology. The study was conducted in accordance with the Declaration of Helsinki. Informed written consent was obtained from the participating members of the families and the parents of the minor children.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yasin, M., Licchetta, L., Khan, N. et al. Genetic heterogeneity in epilepsy and comorbidities: insights from Pakistani families. BMC Neurol 24, 172 (2024). https://doi.org/10.1186/s12883-024-03671-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-024-03671-7