
Abstract
Background
Ischemic stroke in young adults can be caused by a variety of etiologies including the monogenic disorders. Visceral heterotaxy is a condition caused by abnormal left–right determinations during embryonic development. We aimed to determine the cause of a young ischemic stroke patient with visceral heterotaxy.
Case presentation
We performed neurological, radiological, and genetic evaluations in a 17-year-old male patient presenting ischemic stroke and visceral heterotaxy to determine the underlying cause of this rare disease combination. Brain magnetic resonance imaging (MRI) showed evidence of embolic stroke, abdominal computed tomography (CT) showed visceral heterotaxy, and echocardiogram showed cardiac anomaly with right-to-left-shunt (RLS). Whole genome sequencing (WGS) revealed a heterozygous missense variant (NM_018055.5: c.1016 T > C, p.(Met339Val)) in the NODAL gene, which is essential to the determination of the left–right body axis.
Conclusions
Our study highlights the importance of evaluating genetic etiology in young ischemic stroke and the need for stroke risk management in visceral heterotaxy patients with RLS. To the best of our knowledge, we report the first genetically-confirmed case of visceral heterotaxy with young embolic stroke reported to date.
Similar content being viewed by others


Background
Ischemic stroke in young adults is occasionally reported in multiple conditions including monogenic disorders. Genetic testing can find the underlying genetic causes such as defects of coagulation factors, connective tissue abnormalities, or cardiovascular disorders [1, 2]. Despite the impact of monogenic stroke on patients and their families and the importance of assessing genetic causes, patients with young ischemic stroke do not always undergo genetic investigation probably due to their rarity and lack of cost-effective evidence of high-throughput gene sequencing. Efforts have been made to utilize genetic analysis using stroke gene panels [2,3,4] or whole-exome sequencing [5] to manage monogenic stroke, but the detection rate of the causal genes varies and the clinical significance is unclear probably due to the heterogeneous nature of the stroke and lack of our knowledge when monogenic disorder should be suspected in ischemic stroke.
Young adults with congenital cardiac disease have increased risk of cerebral ischemic stroke. While this is primarily due to electro-abnormalities, such as atrial fibrillation, embolic stroke from the venous system due to the right-to-left shunt may also be a factor even in the absence of arrythmia. Therefore, it may be important to consider the preventive medications in managing the patient. However, there has been no study on cardioembolic stroke due to visceral heterotaxy.
Here we report a patient with visceral heterotaxy and embolic stroke whose genetic testing was useful to identify a novel pathogenic variant in the NODAL gene, emphasizing the importance of performing genetic analysis in a patient with young ischemic stroke.
Case presentation
The patient is a 17-year-old male with asplenia syndrome (complete endocardial deficiency, double outlet right ventricle, and pulmonary artery stenosis) as a congenital heart malformation, although his symptoms are only mild cyanosis, and he is independent in activities of daily living (ADL). His parents were healthy and he was an only child. He was referred to our hospital with a sudden episode of transient left hemiparesis. He suddenly became aware of weakness and sensory disturbance on the left upper and lower limbs while defecating. The symptoms gradually improved within 3 h, but the sensory disturbance remained. There was no marked family history and his parents were healthy.
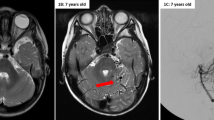
We suspected cerebrovascular disease, because he had a cardiac malformation that could cause a right-to-left shunt. The brain MRI showed cerebral infarction on the right temporo-parietal lobe, and magnetic resonance angiography (MRA) showed no vessel abnormalities (Fig. 1A, B). Based on the findings of MRI, we concluded that the patient had an embolic mechanism through cardiac malformation. Abdominal Computer tomography (CT) scan showed visceral heterotaxy (Fig. 1C). Transthoracic echocardiography revealed endocardial cushion defect (ECD) (Fig. 1D). We were not able to perform transesophageal echocardiography. No atrial fibrillation was reported on standard electrocardiogram (ECG) and Holter ECG.

Clinical and radiological features of the patient. A The head MRI (Diffusion weighted image) shows acute cerebral infarction on temporo-parietal lobe. B MR angiography image shows no vessel abnormality. C Abdominal CT shows visceral heterotaxy. S: Stomach. L: Liver. D Echocardiogram. Echocardiogram shows ECD. RV: Right ventricle. LV: Left ventricle
After admission at the age of 17, blood tests showed slightly low activity of antithrombin III (61%) and protein C (63%), and the presence of congenital coagulopathy was suspected, although genetic testing for SERPINC1 and PROC were negative. In addition, there was no evidence of antiphospholipid antibody syndrome or hyper-homocystinemia, which could cause juvenile cerebral infarction. There were no signs of infectious endocarditis and of deep vein thrombosis (DVT). However, the level of D-dimer FDP was slightly elevated 0.6 μg/ml (normal range: 0–0.5 μg/ml) and the Risk of Paradoxical Embolism (RoPE) score [6] was high (score = 10). Therefore, we diagnosed him as a paradoxical cerebral embolism caused by ECD and potential DVT with right-to-left shunt.
To further investigate the cause of his stroke, whole-genome sequencing (WGS) was performed on the patient (Fig. 2A, See online method). WGS of the patient revealed a novel heterozygous variant NM_018055.5: c.1016 T > C: p.(Met339Thr) (chr10:70,432,964–70432964 in the genomic coordinate of GRCh38) in the NODAL gene (NM_018055.5, MIM#601,265) (Fig. 2B), which is responsible for autosomal dominant visceral heterotaxy (MIM#270,100) [7]. The obtained coverage of WGS was 33.5 x. The unaffected father also had this variant, suggesting incomplete penetrance as reported [7] (Fig. 1A). The father underwent an abdominal CT scan that was completely normal. Sanger-sequencing validated the variant (Fig. 1B). This variant has not been reported in the gnomAD database or the WGS database of 9850 Japanese control individuals [8]. The missense variant was predicted to be damaging with CADD-PHRED score of 26.2. This variant was located in the evolutionally-conserved Nodal active ligand (110 aa) (Fig. 2C, D) and showed high conservation scores (GERP of 5.84, SIFT of 0, PhyloP of 4.603, PolyPhen-2 of 0.998, and PhastCons of 1). MutationTaster prediction was “Disease causing”. Alteration of the same amino acid, c.1015A > G, p.(Met339Val), was reported to cause a mild reduction in nodal signaling by using activin-responsive ARE-luciferase reporter assay in a zebrafish embryo, although detailed clinical description of the patient with the Met339Val variant was unavailable [9]. Protein structure of homodimerized Nodal and its co-receptor CFC1 (CFC1-EGF domain) was modeled by AlphaFold2 [10], which showed that Met339 and other previously reported pathogenic variants were located at the Nodal-CFC1 binding site (Fig. 2E). We further searched for pathogenic variants of the PROC gene in the WGS data that may explain the decrease in Protein C in this patient, but were unable to find any candidate variants. Furthermore, we did not identify any pathogenic variants that were responsible for monogenic stroke panel genes [2].

Genetic analysis of NODAL variant. A Family tree of the patient. WT: wild-type (B) Sanger-sequencing validated c.1016 T > C variant in the patient and the father. C The scheme of NODAL protein. Nodal is post-translationally cleaved and forms mature active ligand (red). Met339 is located in the active ligand (arrow). D Met339 and nearby residues are conserved across species. Genome alignment was obtained from UCSC genome browser (http://genome.ucsc.edu). E Evaluation of protein structure of M394 (red) and other residues (blue) on the TGF-β domain, whose changes are reported to cause heterotaxy. Protein structures of homo-dimerized Nodal mature protein (gray) and CFC1-EGF domain of CFC1 (yellow) were modeled by Alphafold2. L, S and F (orange) in CFC1 represent the three residues reported to be essential for binding to Nodal in CFC1 of mouse Cripto [11]. The molecular structure was drawn by PyMOL (www.pymol.org). Amino acids reported to cause visceral heterotaxy including M339 cluster at the CFC1 and Nodal binding site
Anticoagulation therapy was started with continuous intravenous unfractionated heparin and switched to warfarin. The patient showed no recurrence of ischemic stroke for one and a half years as of now.
Discussion and conclusions
We described a novel NODAL missense variant in a patient presenting with young embolic stroke and visceral heterotaxy. This variant was located in a highly conserved Nodal active domain, and analysis of the protein structure suggested that it may have a role in CFC1 binding. It is noteworthy that the unaffected father has the same variant. Disruption of the left–right axis determination in embryonic development can result in random left–right selection [12]. In support of this, genetic analyses of heterotaxy family showed approximately 50% penetrance, even in monozygotic twins [13]. A heterotaxy family with a NODAL variant was reported, suggesting a ~ 50% penetrance rate [14]. Collectively, it is likely that the unaffected father with Met339Ther did not show heterotaxy due to random determination of the left–right axis. Family-based genetic analysis using high-throughput sequencers usually uses variant filtering based on a hypothesis of 100% penetrance. It may be important to consider this randomness of disease manifestation when performing genetic analysis in a family with visceral heterotaxy.
The patient had an embolic stroke. We considered whether genetic abnormality and cerebral infarction are related. As echocardiography showed an apparent right-to-left shunt, it may be possible that a right-to-left shunt in the heart allowed clots in the venous system to enter the brain circulation and caused an embolic stroke. Although we could not detect DVT, we suspected that the abnormal finding of protein C level inspires the insidious occurrence of DVT. Although the patient showed a mildly reduced Protein C level (63%), no pathogenic variants were found in the PROC gene. It is reported that protein C deficiency due to pathogenic genetic variants can decrease Protain C level below 55%. Mild reductions (55–65%) are also seen in healthy individuals [15]. It may be caused by less effective genetic conditions, such as common eQTL variants in regulatory regions [16], although we could not draw any conclusions about this possibility. Thus, we diagnosed the type of cerebral infarction as paradoxical cerebral embolism. We should be aware that patients with a NODAL variant may develop a stroke. Although not all young ischemic stroke patients necessarily require genetic testing, patients with possible monogenic diseases observed in the present case should be considered for obtaining the correct diagnosis and providing genetic counseling.
In conclusion, we report a novel NODAL variant in a young embolic stroke patient with visceral heterotaxy. It would be important to investigate a monogenic disorder that may be secondarily related to a young embolic stroke. It is also important to carefully evaluate unaffected family members when performing family-based WGS, as heterotaxy can have a penetrance of 50% due to random left–right selection.
Availability of data and materials
WGS data is private and unavailable.
References
Ekker MS, Boot EM, Singhal AB, et al. Epidemiology, aetiology, and management of ischaemic stroke in young adults. Lancet Neurol. 2018;17:790–801.
Ilinca A, Puschmann A, Putaala J, et al. Updated stroke gene panels: rapid evolution of knowledge on monogenic causes of stroke. Eur J Hum Genet. 2023;31:239–42.
Tan RYY, Traylor M, Megy K, et al. How common are single gene mutations as a cause for lacunar stroke? A targeted gene panel study. Neurology. 2019;93:e2007–20.
Fang F, Xu Z, Suo Y, et al. Gene panel for Mendelian strokes. Stroke Vasc Neurol. 2020;5:416–21.
Ilinca A, Martinez-Majander N, Samuelsson S, et al. Whole-exome sequencing in 22 young ischemic stroke patients with familial clustering of stroke. Stroke. 2020;51:1056–63.
Kent DM, Ruthazer R, Weimar C, et al. An index to identify stroke-related vs incidental patent foramen ovale in cryptogenic stroke. Neurology. 2013;81:619–25.
Gebbia M, Ferrero GB, Pilia G, et al. X-linked situs abnormalities result from mutations in ZIC3. Nat Genet. 1997;17:305–8.
Kawai Y, Watanabe Y, Omae Y, Miyahara R, Khor SS, Noiri E, Kitajima K, Shimanuki H, Gatanaga H, Hata K, et al. Exploring the genetic diversity of the Japanese population: Insights from a large-scale whole genome sequencing analysis. PLoS Genet. 2023;19:e1010625. https://doi.org/10.1371/journal.pgen.1010625.
Roessler E, Pei W, Ouspenskaia MV, et al. Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephaly. Mol Genet Metab. 2009;98:225–34.
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Yan YT, Liu JJ, Luo Y, et al. Dual roles of Cripto as a ligand and coreceptor in the nodal signaling pathway. Mol Cell Biol. 2002;22:4439–49.
Layton WM Jr. Random determination of a developmental process: reversal of normal visceral asymmetry in the mouse. J Hered. 1976;67:336–8.
Noone PG, Bali D, Carson JL, et al. Discordant organ laterality in monozygotic twins with primary ciliary dyskinesia. Am J Med Genet. 1999;82:155–60.
Li AH, Hanchard NA, Azamian M, et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur J Hum Genet. 2019;27:563–73.
Miletich J, Sherman L, Broze G Jr. Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med. 1987;317:991–6.
Gudjonsson A, Gudmundsdottir V, Axelsson GT, et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat Commun. 2022;13:480.
Acknowledgements
We are grateful to the patient and his parents for participating in our study. We would like to thank to Ritsuko Oikawa and Yoshiko Miyake for their technical assistance and Yoshikazu Haramoto for his comments about Nodal. Additionally, we are thankful for the partial support provided by the supercomputer at ROIS National Institute of Genetics for computation.
Funding
This work was supported by grants from the Japan Agency for Medical Research and Development (AMED No.22ek0109493). We thank the patient and his parents for participating in our study. Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Author information
Authors and Affiliations
Contributions
KK and SM wrote the main manuscript and prepared all figures. NM, YK, YM, and KT contributed to obtaining WGS data. YK and SM analyzed and interpreted the WGS data. KK, YK, KT, HA, YY, and TS contributed to obtaining and interpreting clinical data. YY and SM contributed to the conceptualization of the study. All authors read and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed and disclosure consents were obtained from the patient and a parent for study participation. Institutional review board of St. Marianna University School of Medicine approved the study protocol (#4983).
Consent for publication
Consent for publication was obtained from the patient and a parent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kaburagi, K., Hagiwara, Y., Tachikawa, K. et al. A novel NODAL variant in a young embolic stroke patient with visceral heterotaxy. BMC Neurol 24, 119 (2024). https://doi.org/10.1186/s12883-024-03619-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-024-03619-x