
Abstract
Background
Chronic progressive external ophthalmoplegia (CPEO) is a mitochondrial disease with slowly progressive bilateral ptosis and symmetric ophthalmoplegia due to a genetic mutation that results in defective oxidative phosphorylation. Common genes that are implicated in CPEO include POLG, RRM2B, ANT1 and PEO1/TWNK. Here, we report a case of a patient diagnosed with CPEO caused by a novel mutation in PEO/TWNK after suffering a right pontine stroke.
Case presentation
A 70-year-old man with history of chronic progressive bilateral ptosis and ophthalmoplegia, as well as similar ocular symptoms in his father and grandfather, presented with acute onset of right hemifacial weakness and dysarthria. Brain MRI revealed an acute ischemic stroke in the right dorsal pons. The patient did not experience diplopia due to severe baseline ophthalmoplegia. Creatine kinase was elevated to 6,080 U/L upon admission and normalized over the course of one week; electromyography revealed a myopathic process. Genetic testing revealed a novel mutation c.1510G > A (p. Ala504Thr) in a pathogenic “hot spot” of the C10ORF2 gene (TWNK/PEO1), which is associated with CPEO. The mutation appears to be deleterious using several pathogenicity prediction tools.
Conclusions
This case report describes a patient with late-onset CPEO caused by a novel, likely pathogenic, mutation in the TWNK gene. Although the patient presented with a pontine stroke, it manifested with solely new onset facial palsy, as he had a severe underlying ophthalmoplegia secondary to his CPEO.
Similar content being viewed by others


Background
Chronic progressive external ophthalmoplegia (CPEO) represents a heterogenous spectrum of mitochondrial disorders that manifest clinically as slowly progressive bilateral ptosis and symmetric ophthalmoplegia. In some cases, weakness of the pharyngeal muscles can cause dysphagia. CPEO can present as an isolated myopathy or in conjunction with other neurologic symptoms such as hearing loss, neuropathy, and ataxia (termed CPEO +) [1]. The disease is more common in females (ratio 2.5:1), and it typically presents during mid-adulthood [2]. CPEO results from defective oxidative phosphorylation in mitochondria caused by: 1) mutations in nuclear DNA responsible for maintenance of mitochondrial DNA (mtDNA), 2) point mutations in mitochondrial transfer RNA genes, or 3) single, large deletions of mtDNA [1,2,3,4,5,6].
The nuclear genes associated with CPEO are POLG/POLG2, PEO1 (TWNK), RRM2B, SLC25A4 (ANT1), DNA2, OPA1, TYMP, MGM1, RNASEH1, TK2, and DGUOK; mutations in these genes are usually inherited in an autosomal dominant or autosomal recessive fashion [1,2,3,4, 7, 8]. The mitochondrial transfer RNA gene implicated in CPEO is MT-TL1; mutations in this gene are inherited in a mitochondrial pattern. Single, large deletions of mtDNA are typically sporadic; these sporadic events make up approximately half of the cases [1, 3]. Here, we report a patient presenting with CPEO caused by a novel mutation in PEO1/TWNK, who was diagnosed after he presented with right facial palsy due to a pontine stroke.
Case presentation
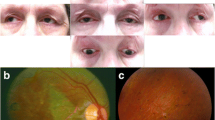
A 70-year-old African American male presented to the emergency room after acute onset of right facial weakness and dysarthria for five hours. Demographic data is available in Table 1. On further questioning, he had non-fluctuating, progressive ptosis for approximately 10 years, as well as worsening dysphagia for liquids and solids over the previous several months. There was a family history of bilateral ptosis in his father and paternal grandfather. Detailed examination demonstrated symmetrical and bilaterally reactive pupils, severe bilateral ptosis and ophthalmoplegia (Fig. 1), with complete vertical gaze palsy and severely limited horizontal eye movements, and right-sided facial weakness involving the orbicularis oculi and oris. Convergence was impaired. Palatal elevation and swallowing were impaired. There was a complete vertical gaze palsy. There was no muscle weakness in the upper or lower limbs. Deep tendon reflexes were normal aside from absent ankle jerks. The patient denied pain.

Bilateral ophthalmoplegia and ptosis. Severely restricted bilateral eye movement on rightward gaze (A), and leftward gaze (B); severe bilateral ptosis requiring physical lifting of eyelids in order for the patient to see (C-D)
Based on acuity of symptoms, an acute stroke was considered. A brain CT scan was obtained in the emergency department and was negative for hemorrhage. A brain MRI the following day revealed an acute ischemic stroke in the right dorsal pons at the level of the seventh cranial nerve nucleus and tract (Fig. 2). The patient presented out of the thrombolysis window, so the ischemic stroke was managed with secondary prevention measures. Initial relevant lab work (Table 1) revealed elevated creatine kinase (CK) to 6,080 U/L (decreased to 477 U/L over a week), borderline high low-density lipoprotein (LDL) (131 mg/dL), and normal hemoglobin A1c (5.6%).

Pontine stroke on MRI Brain and diagram of affected and nearby structures. Axial sections through the pons demonstrate a lesion which was hyperintense on diffusion-weighted imaging (DWI) sequence (A), hypointense on apparent diffusion coefficient (ADC) sequence (B), and hyperintense on T2 and fluid attenuated inversion recovery (FLAIR) (C). The approximate location of facial and abducens nerves and related oculomotor structures are shown in image D. V: ventricle, CN: cranial nerve, PPRF: parapontine reticular formation, MLF: medial longitudinal fasciculus
A nerve conduction study, in conjunction with absent ankle reflexes, suggested a possible length-dependent sensory axonal neuropathy (Table 2). The compound muscle action potentials (CMAPs) were single-peaked. Repetitive nerve stimulation of the left facial nerve (recorded at nasalis) and left spinal accessory nerve (recorded at trapezius) did not show any decrement of the CMAP amplitudes. Neither oral pyridostigmine nor an ice pack test resulted in improvement of oculobulbar symptoms. Serum antibodies against acetylcholine receptors and muscle specific tyrosine kinase were negative.
Needle electromyography (EMG) showed fibrillation potentials and positive waves in all muscles tested in the right upper and lower limb and thoracic paraspinals; motor unit action potentials were overall normal and motor unit recruitment was normal in all muscles except the right deltoid, which was myopathic. Cerebrospinal fluid (CSF) analysis showed normal white blood cell count and protein levels and negative cytology. The following tests were negative or normal: CT scan of chest/abdomen/pelvis, serum and urine protein immunoelectrophoresis and immunofixation, and serum and CSF lactate and angiotensin converting enzyme levels. The patient declined a muscle biopsy.
EMG Summary Table | |||||||||
|---|---|---|---|---|---|---|---|---|---|
Spontaneous | MUAP | Recruitment | |||||||
IA | Fib | PSW | Fasc | Other | Amp | Dur | Morp | Pattern | |
R. DELTOID | Inc | None | 1 + | None | None | N | N | N | early |
R. BICEPS | N | 1 + | 1 + | None | None | N | N | N | N |
R. FLEXOR CARPI RADIALIS | Inc | 1 + | 1 + | None | None | N | N | N | N |
R. 1ST DORSAL INTEROSSEOUS | Inc | 1 + | None | None | None | N | N | N | N |
R. TIBIALIS ANTERIOR | Inc | 1 + | 1 + | None | None | N | N | N | N |
R. VASTUS MEDIALIS | Inc | None | 1 + | None | None | N | N | N | N |
R. THORACIC PARASPINALS | Inc | 2 + | 2 + | None | None | ||||
Based on the suspicion of a mitochondrial disorder and the absence of a viable tissue biopsy for biochemical and histochemical analysis, genetic testing was completed in the blood. Whole-exome sequencing (WES), as opposed to multi-gene panel or targeted next-generation sequencing approaches, was pursued given its emergence as a preferred and more effective diagnostic tool for suspected mitochondrial disease and other mimic neuromuscular disorders such as congenital myasthenias [9,10,11]. The success of WES compared to other approaches stems from the significant genetic and clinical heterogeneity of mitochondrial disorders and of neuromuscular disorders that can have overlap** clinical features [9].
WES demonstrated a variant (c.1510G > A (p.Ala504Thr) in the PEO1/TWNK (C10ORF2, NM_021830.4/5) gene, which encodes a mitochondrial protein known as “twinkle,” a DNA helicase involved in maintaining mtDNA [12]. The p.Ala504Thr substitution is likely deleterious using several in-silico pathogenicity prediction tools: Sorting Intolerant From Tolerant (SIFT) [13], PolyPhen2 [14], Align Gradient Validation Gradient Deviation (GVGD) [15], and Rare Exome Variant Ensemble Learner (REVEL) [16]. As such, this mutation represents a novel mutation associated with CPEO.
After the patient was discharged from the hospital, he was lost to follow-up; and genetic testing could not be completed on any of the patient’s affected family members as they were deceased.
Discussion and conclusions
Clinically, this patient’s presentation of both an acute pontine stroke and CPEO presents several interesting learning points. First, it is rare for a pontine stroke to cause an isolated CN VII palsy. If the patient did not have baseline ophthalmoplegia, his stroke would have caused impaired right eye abduction due to right abducens palsy, impaired right eye adduction due to involvement of right medial longitudinal fasciculus, and impaired left eye adduction due to involvement of parapontine reticular formation (i.e. one and a half syndrome). In addition to one and a half syndrome, the stroke would have also caused a right facial palsy due to involvement of CN VII, as shown in Fig. 2D [17]. However, his stroke-related oculomotor symptoms were obscured by his underlying ophthalmoplegia. Second, stroke or stroke-like episodes are potential manifestations of mitochondrial disease, classically in the context of mitochondrial encephalomyopathy, lactic acidosis, and stroke like symptoms (MELAS). Our patient did not have the MELAS phenotype or a known mutation associated with MELAS, and the location of stroke was uncharacteristic for strokes associated with MELAS, which are typically cortical or thalamic and may not conform to any known vascular territory [18]. On the other hand, it has been proposed that mitochondrial disease aside from MELAS may predispose to stroke [19]. Considering his age, presence of small vessel disease and hypertensive angiopathy on brain imaging and range of elevated blood pressures while admitted, the etiology of his pontine lacunar stroke was likely small vessel disease. Computed Tomography Angiography (CTA) of the head and neck demonstrated diffuse atherosclerotic disease, including multifocal stenosis of internal carotid arteries, vertebral arteries and bilateral middle cerebral arteries. Based on time course, the constellation of presenting symptoms were separated based on acuity. The patient experienced sudden-onset right hemifacial paralysis and dysarthria. The weakness of upper and lower face in this patient suggested a lower motor neuron lesion, involving the facial nerve nucleus, fasciculus or nerve. A CVA involving cranial nerve VII nucleus or its tract was considered, given the acute onset and age of the patient.
The differential diagnoses for this patient’s progressive symmetric oculobulbar weakness included: 1) neuromuscular junction (NMJ) disorders, specifically myasthenia gravis and congenital myasthenic syndromes; and 2) heredofamilial myopathies, such as CPEO, oculopharyngeal muscular dystrophy, and myotonic dystrophy. In this patient, a neuromuscular junction disorder was considered unlikely based on negative repetitive nerve stimulation, ice pack test, and myasthenia gravis serologies. Normal configuration of CMAPs argued against a slow channel syndrome. The presence of diffuse fibrillations and positive waves in the EMG were suggestive of an underlying myopathy (rather than diffuse denervation) given the overall clinical presentation, markedly elevated CK, and presence of myopathic motor unit recruitment in the deltoid.
A mitochondrial myopathy explained not only the chronic oculobulbar symptoms but also the distal neuropathy and subclinical episode of rhabdomyolysis [20, 21]. Other heredofamilial myopathies, such as oculopharyngeal muscular dystrophy and myotonic dystrophy type I, were excluded with genetic testing. Although a muscle biopsy was not pursued in this case, the clinical characteristics are quite suggestive of a diagnosis of CPEO and align with the effects of similar mutations in the PEO1/TWNK gene.
PEO1 (TWNK) is a nuclear gene that encodes for the twinkle protein and mutations in this gene are associated with CPEO. Autosomal dominant mutations in PEO1 have been shown to cause a constellation of symptoms including ptosis, external ophthalmoplegia, and myopathy. Autosomal recessive mutations in PEO1 have been implicated in infantile-onset spinocerebellar ataxia [22]. Both the clinical presentation and family history (similar symptoms present in patient’s father and paternal grandfather) are consistent with an autosomal dominant inheritance pattern; therefore, we did not proceed with mitochondrial DNA sequencing. The twinkle protein consists of 684 amino acids with a molecular mass of 77kD [12]. Van Hove et al. noted that the protein has three functional domains: a 5’ primase domain, a linker domain, and a 3’ helicase domain [22]. The majority of human mutations occur in the linker and helicase domains, and the pathogenic mutations in PEO1 are clustered between residues Arg303 and Tyr508 [6, 22]. This patient’s novel mutation (p.Ala504Thr) is located in the helicase domain and affects residues that are predicted to be involved with stabilizing oligomeric structure [23, 24].
Our case study has several limitations: 1) we were unable to obtain a muscle biopsy, which may have further supported the diagnosis of a mitochondrial myopathy, and 2) genetic testing could not be performed in the patient’s family members to confirm the familial inheritance and pathogenic nature of the mutation.
To summarize, we present a 70-year-old male with acute onset right hemifacial weakness superimposed on chronic progressive bilateral ptosis, ophthalmoplegia, and dysphagia. The time course of the symptoms and proper anatomic localization were integral in recognizing the presence of multiple pathologies: a pontine stroke with an acute onset, isolated cranial nerve VII palsy, and an underlying CPEO, a mitochondrial myopathy with chronic and progressive oculobulbar weakness. Genetic testing revealed a novel variant in a pathogenic “hot spot” of the PEO1 gene, supportive of the diagnosis of CPEO.
Availability of data and materials
The principal data gathered and analyzed during this study are included in this published article. Data are available from the corresponding author on reasonable request.
Abbreviations
- CPEO:
-
Chronic Progressive External Ophthalmoplegia
- DNA:
-
Deoxyribonucleic Acid
- RNA:
-
Ribonucleic Acid
- mtDNA:
-
Mitochondrial Deoxyribonucleic Acid
- CT:
-
Computed Tomography
- MRI:
-
Magnetic Resonance Imaging
- CK:
-
Creatine Kinase
- LDL:
-
Low-density Lipoprotein
- DWI:
-
Diffusion-Weighted Imaging
- ADC:
-
Apparent Diffusion Coefficient
- FLAIR:
-
Fluid Attenuated Inversion Recovery
- V:
-
Ventricle
- CN:
-
Cranial Nerve
- PPRF:
-
Parapontine Reticular Formation
- MLF:
-
Medial Longitudinal Fasciculus
- CMAP:
-
Compound Muscle Action Potential
- EMG:
-
Electromyography
- CSF:
-
Cerebrospinal Fluid
- WES:
-
Whole-exome sequencing
- SIFT:
-
Sorting Intolerant From Tolerant
- GVGD:
-
Align Gradient Validation Gradient Deviation
- REVEL:
-
Rare Exome Variant Ensemble Learner
- MELAS:
-
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke Like Symptoms
- CVA:
-
Cerebrovascular Accident
- NMJ:
-
Neuromuscular Junction
- kD:
-
Kilodalton
References
McClelland C, Manousakis G, Lee MS. Progressive External Ophthalmoplegia. Curr Neurol Neurosci Rep. 2016;16(6):53.
Heighton JN, Brady LI, Newman MC, Tarnopolsky MA. Clinical and demographic features of chronic progressive external ophthalmoplegia in a large adult-onset cohort. Mitochondrion. 2019;44:15–9.
Rodríguez-López C, García-Cárdaba LM, Blázquez A, Serrano-Lorenzo P, Gutiérrez-Gutiérrez G, San Millán-Tejado B, et al. Clinical, pathological and genetic spectrum in 89 cases of mitochondrial progressive external ophthalmoplegia. J Med Genet. 2020;57(9):643–6.
Hou Y, Zhao X, **e Z, Yu M, Lv H, Zhang W, et al. Novel and recurrent nuclear gene variations in a cohort of Chinese progressive external ophthalmoplegia patients with multiple mtDNA deletions. Mol Genet Genomic Med. 2022;10(5): e1921.
Karanjia R. Chronic progressive external ophthalmoplegia. Acta Ophthalmologica. 2022;100(S275). Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1755-3768.2022.15322. [cited 2023 Apr 9].
Bermejo-Guerrero L, de Fuenmayor-Fernández de la Hoz CP, Serrano-Lorenzo P, Blázquez-Encinar A, Gutiérrez-Gutiérrez G, Martínez-Vicente L, et al. Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. J Clin Med. 2022;11(1):22.
Caporali L, Bello L, Tagliavini F, La Morgia C, Maresca A, Di Vito L, et al. DGUOK recessive mutations in patients with CPEO, mitochondrial myopathy, parkinsonism and mtDNA deletions. Brain. 2018;141(1): e3.
Kierdaszuk B, Kaliszewska M, Rusecka J, Kosińska J, Bartnik E, Tońska K, et al. Progressive External Ophthalmoplegia in Polish Patients—From Clinical Evaluation to Genetic Confirmation. Genes. 2021;12(1):54.
Theunissen TEJ, Nguyen M, Kamps R, Hendrickx AT, Sallevelt SCEH, Gottschalk RWH, et al. Whole Exome Sequencing Is the Preferred Strategy to Identify the Genetic Defect in Patients With a Probable or Possible Mitochondrial Cause. Front Genetics. 2018;9. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2018.00400. [cited 2023 Feb 7].
Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, et al. Use of Whole-Exome Sequencing to Determine the Genetic Basis of Multiple Mitochondrial Respiratory Chain Complex Deficiencies. JAMA. 2014;312(1):68–77.
Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38(3):437–43.
Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28(3):223–31.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34(5):1317–25.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99(4):877–85.
Evans MRB, Weeks RA. Putting pontine anatomy into clinical practice: the 16 syndrome. Pract Neurol. 2016;16(6):484–7.
Cheng W, Zhang Y, He L. MRI Features of Stroke-Like Episodes in Mitochondrial Encephalomyopathy With Lactic Acidosis and Stroke-Like Episodes. Front Neurol. 2022;13: 843386.
Anderson CD, Biffi A, Rahman R, Ross OA, Jagiella JM, Kissela B, et al. Common mitochondrial sequence variants in ischemic stroke. Ann Neurol. 2011;69(3):471–80.
Nance JR, Mammen AL. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve. 2015;51(6):793–810.
de Barcelos IP, Emmanuele V, Hirano M. Advances in Primary Mitochondrial Myopathies (PMM). Curr Opin Neurol. 2019;32(5):715–21.
Van Hove JLK, Cunningham V, Rice C, Ringel SP, Zhang Q, Chou PC, et al. Finding twinkle in the eyes of a 71-year-old lady: A case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am J Med Genetics Part A. 2009;149A(5):861–7.
Ziebarth TD, Farr CL, Kaguni LS. Modular Architecture of the Hexameric Human Mitochondrial DNA Helicase. J Mol Biol. 2007;367(5):1382–91.
Riccio AA, Bouvette J, Perera L, Longley MJ, Krahn JM, Williams JG, et al. Structural insight and characterization of human Twinkle helicase in mitochondrial disease. Proc Natl Acad Sci. 2022;119(32): e2207459119.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YE: Data analysis, draft of original manuscript, manuscript revisions, and figure creation; KR: study conceptualization, figure creation, manuscript revisions, and study supervision; CMG: participated in the clinical care of this patient and helped obtain and generate the clinical data utilized, manuscript revisions, and study supervision; KS: participated in the clinical care of this patient and helped obtain and generate the clinical data utilized, study conceptualization, data analysis, manuscript revisions, and study supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This case study was investigated in accordance with the ethical principles outlined in the Declaration of Helsinki. The patient provided written informed consent to participate in this study and have its contents published.
Consent for publication
The patient provided written informed consent to participate in this study and have its contents published. The patient’s written consent is available per request.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eliyan, Y., Rezania, K., Gomez, C.M. et al. Pontine stroke in a patient with Chronic Progressive External Ophthalmoplegia (CPEO): a case report. BMC Neurol 23, 231 (2023). https://doi.org/10.1186/s12883-023-03249-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-023-03249-9