
Abstract
Background
Thrombotic thrombocytopenic purpura (TTP) is rare and severe thrombotic microangiopathy characterized by thrombocytopenia, hemolytic anemia, and renal dysfunction. In contrast, essential thrombocythemia (ET) is a myeloproliferative disease associated with an abnormal increase in platelet numbers. Previous studies reported several cases of the development of ET in patients with TTP. However, the case of an ET patient complicated with TTP has not been previously reported. In this case study, we present a patient with TTP who was previously diagnosed with ET. Therefore, to the best of our knowledge, this is the first report of TTP in ET.
Case presentation
A 31-year-old Chinese female who was previously diagnosed with ET presented with anemia and renal dysfunction. The patient had been on long-term treatment with hydroxyurea, aspirin, and alpha interferon (INF-α) for ten years. The diagnosis of TTP was confirmed by clinical features, schistocytes noted on the peripheral blood smear, and lower ADAMTS13 activity (8.5%), together with the renal biopsy results. INF-α was discontinued, and the patient was then treated with plasma exchange and corticosteroids. After one year of follow-up, the patient had a normal hemoglobin level and platelet numbers, and her ADAMTS13 activity had improved. However, the patient’s renal function remains impaired.
Conclusions
We report a case of an ET patient complicated with TTP that was possibly due to INF-α, highlighting the potential complications associated with long-term ET therapy. The case also highlights the importance of considering TTP in patients with pre-existing ET who present with anemia and renal dysfunction, extending the spectrum of known studies.
Similar content being viewed by others


Background
Thrombotic thrombocytopenic purpura (TTP) and essential thrombocythemia (ET) are two hematologic disorders characterized by apparently opposite clinical features. TTP is a rare and severe life-threatening disease commonly defined by thrombocytopenia (platelet count less than 100×109/L), microangiopathic hemolytic anemia (MAHA), neurologic changes, renal dysfunction, and fever [1]. It is caused by severely reduced activity of the von Willebrand factor-cleaving protease ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13). In contrast, ET was classified as a myeloproliferative neoplasm by Damesheck in 1951. According to the World Health Organization, ET is identified by a sustained elevation in platelet numbers (more than 450×109/L) and megakaryocytic hyperplasia in the bone marrow [2]. Driver mutations, such as JAK2V617F (55%), CALR (20%-25%), and MPL (5%), are also often associated [3]. The primary complications in ET are thrombohemorrhagic events and leukemic/fibrotic transformation. Renal involvement by ET is rare. However, renal failure may develop in patients with ET due to bilateral thrombosis of the renal arteries or occlusion of the urinary tract by blood clots [4, 5]. Here, we report a patient with ET who developed anemia and renal dysfunction after treatment with alpha interferon (INF-α).
Case presentation
A 31-year-old female patient was admitted to the Department of Nephrology at West China Hospital after experiencing edema and fatigue for approximately seven days. She did not have a fever, diarrhea, purpura, or dark urine. The patient had a ten-year history of ET and was prescribed hydroxyurea, aspirin, and INF-α. The patient received a dose of interferon of 3×106 IU/week for the past five years and received the latest interferon treatment four days before arriving at our unit. The patient’s blood pressure and blood tests were monitored every three months throughout her interferon treatment, including liver enzymes, complete blood count, and renal function, none of which were abnormal. Her personal, family, and psychosocial histories were unremarkable. Upon physical examination, her temperature was 36.5 ℃, heart rate was 75 beats per minute, respiratory rate was 20 breaths per minute, and blood pressure was 140/90 mmHg. The patient’s weight was 55 kg, and her height was 158 cm. No skin rash or bleeding spots were seen, but moderate edema was observed on the face and extremities. The muscle strength and muscle tone of the four limbs were normal, and no abnormalities were identified in the physical examination of the lung, heart, and abdomen. Laboratory test results revealed anemia with schistocytosis, elevated lactic dehydrogenase (LDH) and creatinine levels, and a negative Coomb's tests, raising concern for thrombotic microangiopathy (TMA) (Table 1). Thus, hydroxyurea and INF-α were discontinued, and prednisone at 1 mg/kg/day was started.
A central venous catheter was placed due to a further increase in creatinine (529 umol/L) and oliguria, and hemodialysis combined with single-volume plasma exchange (PE) was conducted after obtaining a blood sample for the ADAMTS 13 activity test. Given the patient’s history of ET, leukemic or fibrotic disease transformation was a leading consideration in this case. Autoimmune diseases and infections were also suspected as possible causes. However, the bone marrow biopsy showed proliferation mainly of megakaryocyte lineage cells, with increased numbers of enlarged, mature megakaryocytes with hyper-lobulated nuclei. Tests for tumor markers, antinuclear antibody, extractable nuclear antigens, anticardiolipin antibodies, antineutrophil cytoplasmic autoantibody, complement C3, complement C4, complement factor B, human immunodeficiency virus antibody test, hepatitis B virus DNA, Epstein-Barr virus DNA, and septic work-up, including blood, urine, and stool, were negative. The coagulation assay and fibrinogen test results were also within the normal ranges. We did not observe any typical findings of malignant hypertension on her optic fundi. The patient’s ADAMTS 13 activity was 8.5%, and brain computed tomography scan and ultrasound examinations of the kidneys did not reveal any abnormalities. The renal biopsy showed a double contour of the basement membrane and arteriole with luminal narrowing. Immunofluorescence tests for IgG, IgA, IgM, C3, C4, and C1q were all negative. The electron micrograph showed diffuse endothelial swelling with obliteration of the capillary lumina. A diagnosis of TMA was made based on the pathological examination (Fig. 1).

Characteristic histological findings in the renal biopsy. Periodic acid-methylamine silver staining shows a double-contour appearance of the basement membrane (red arrows) and an arteriole with luminal narrowing due to intimal edema and widening (yellow arrow)
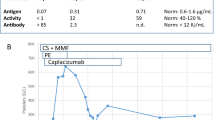
Following five cycles of PE and hemodialysis, the patient exhibited improved clinical symptoms, including elevated hemoglobin level, normalized platelet count, and decreased LDH level (Fig. 2). PE was then paused after 12 cycles. By day 66, the repeated ADAMTS 13 test showed that the activity had increased to 35.8%, and the platelet count had risen to 563×109/L. Then, hydroxyurea was added to the treatment regimen. On day 75, the patient was discharged in stable condition. Unfortunately, despite the stable results, renal function had not recovered, and she still required continuous hemodialysis during her one-year follow-up.

Dynamic changes in platelet counts and lactate dehydrogenase (LDH) levels during hospitalization. The plasma exchange (PE) dates are denoted by blue arrows and hydroxyurea administration is denoted by a thin, black arrow
Discussion and conclusions
As ET and TTP have distinct natural histories and require different treatment modalities, it is crucial to acknowledge that the two diseases may present sequentially. A published case described the development of ET in a patient with TTP [6]. However, there have been no reported cases of ET patients complicated with TTP. To our knowledge, this was the first documented report of a patient with both ET and TTP. This case highlights the complexity and variability of platelet disorders and emphasizes the importance of reviewing peripheral blood smears, ADAMTS13 activity to evaluate TTP, and bone marrow aspirations to diagnose thrombocythemia accurately.
In our case, the renal biopsy revealed TMA, which commonly encompasses hemolytic uremia syndrome (HUS) and TTP. Typical HUS accounts for 90% of HUS cases and is caused by Shiga toxin-producing organisms Escherichia coli and Shigella dysenteriae [7]. Typical HUS predominantly affects children and has a classic prodrome of abdominal pain with diarrhea. In contrast, atypical HUS, which lacks prodromal symptoms, is caused by complement dysregulation and can be triggered by an upper respiratory tract infection [8]. Our patient was an adult with no prodrome of abdominal pain or diarrhea, and no significant abnormality was found in complement tests or fecal culture, reducing the possibility of HUS.
TTP, first described by Moschcowitz in 1924, is a life-threatening form of TMA characterized by systemic microvascular platelet aggregation and erythrocyte destruction. Previous studies showed that ADAMTS13 activity below 10% was specific for TTP and could be used to differentiate TTP from HUS and other TMAs [9]. The incidence of TTP is 2.2/1,000,000 per year, with a prevalence of 13-20/1,000,000, and is more common in women [10, 11]. Untreated TTP has a high mortality rate. However, with proper treatment, the 30-day mortality rate can be reduced to 4 – 7% [12]. Some direct deaths can be attributed to delayed diagnosis and, thus, postponed treatment [13]. Given the risk of acute disseminated microthrombus formation, irreversible organ damage, and death, it is critical to initiate treatment in patients with suspected TTP. PE is initiated once the diagnosis of TTP is suspected, acting as the cornerstone of immediate treatment [14, 15]. In our case, the patient presented with two symptoms of classic TTP pentalogy (MAHA and renal failure) and an ADAMTS13 activity of 8.5%. Therefore, after discontinuing INF-α and hydroxyurea, we immediately treated the patient with PE and corticosteroid (1 mg/kg/d).
TTP could be caused by a variety of factors, including infection, cancer, autoimmune diseases (such as systemic lupus erythematous, Gougerot-Sjögren syndrome, and antiphospholipid syndrome), organ transplantation, pregnancy, and drugs [1]. In this particular case, the patient had ET, but her bone marrow histopathology showed a stable condition without signs of leukemic or fibrotic disease transformation. The patient also did not exhibit any symptoms of autoimmune diseases (arthralgia or rash), and the related laboratory tests (antinuclear antibody, anticardiolipin antibody, antineutrophil cytoplasmic autoantibody, and Coomb's test) were all negative. The patient showed no signs of infection. Therefore, drug-induced TTP was suspected based on these factors.
Drug-induced TTP has been recognized in the last few decades, accounting for about 12% of TTP cases [16]. However, the majority of drug-induced TTP case reports are challenging to interpret because of the ambiguity surrounding the relationship between drug exposure and the onset of TTP [17]. The most common drugs associated with TTP are ticlopidine, quinine, clopidogrel (immune-mediated reaction), mitomycin C, and cyclosporine (dose-related toxicity) [17]. In this report, our patient had been treated with hydroxyurea [18, 19] and INF-α [20], both of which have been reported to cause TTP. However, retreatment with hydroxyurea did not worsen the MAHA symptoms, and her condition became more stable. Therefore, it was improbable that hydroxyurea was the drug responsible for inducing TTP. A causality assessment using the Naranjo algorithm was also performed [21]. The patient scored 6 points on the algorithm for INF-α (a score of “probable” on the probability scale), indicating that INF-α was probably the drug causing TTP.
Due to its antineoplastic, antiviral, and immunomodulatory properties, INF-α has been extensively used in the treatment of various human diseases [22]. It is actively used in the treatment of hematological malignancies, such as myeloproliferative neoplasms [22]. INF-α also plays a prominent role in the treatment of viral syndromes, such as hepatitis C [23]. However, despite its beneficial therapeutic properties, INF-α therapy has been associated with several well-documented toxicities [24]. Previously published cases described TTP as a side effect of INF-α in patients with viral hepatitis, chronic myelogenous leukemia, and polycythemia vera [25,26,27,28]. These cases emphasized a causal relationship between INF-α therapy and TMA [20]. To our knowledge, TTP associated with INF-α has not been reported in patients with ET. The characteristics of TTP in our case were different from other TTP patients that used INF-α. In our case, the patient's platelet count was not significantly reduced (platelet count was not less than 100×109/L), which may be related to the patient’s ET. Diagnosing TTP clinically was challenging as the patient's platelet level did not show a significant reduction. TTP also complicated the treatment of ET and required better doctor-patient coordination.
The primary treatment for ET aims to prevent thrombotic complications. Aspirin therapy or observation alone is effective for lower-risk patients, while cytoreductive therapy is reserved for patients with high-risk disease [29]. Moreover, when TTP occurs, cytoreductive therapy is discontinued, and PE and corticosteroid therapy are initiated immediately. Patients require long-term dynamic monitoring by peripheral blood smears, ADAMTS13 activity, and other tests so that doctors can adjust the treatment plan and maintain ET and TTP in a relatively stable state.
Drug-induced TTP can occur through various mechanisms, including toxic dose-related reactions, immune reactions, and metabolism-mediated mechanisms [30]. However, the exact mechanisms underlying INF-α-associated TTP remain unclear. Some studies suggested that INF-α could cause direct damage to endothelial cells by decreasing vascular endothelial growth factor production or generating an ADAMTS-13 inhibitor through T-cell activation [20, 31]. Recent research on TTP pathogenesis identified a common pathway of complement activation in all TTP patients [32]. Further studies are necessary to elucidate the mechanism of TTP induced by INF-α.
In conclusion, we report the case of a patient with ET who developed TTP that was possibly due to INF-α. PE might help to control disease activity, but further research is needed for a better understanding of the mechanisms underlying this rare complication and the future detection of TTP in a timely manner.
Availability of data and materials
All data used to support the findings of this study are included in the article.
Abbreviations
- TTP:
-
Thrombotic thrombocytopenic purpura
- ET:
-
Essential thrombocythemia
- MAHA:
-
Microangiopathic hemolytic anemia
- LDH:
-
Lactic dehydrogenase
- TMA:
-
Thrombotic microangiopathy
- PE:
-
Plasma exchange
- HUS:
-
Hemolytic uremia syndrome
References
Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129:2836–46.
Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8:15.
Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112:141–9.
Bello Nicolau I.Conde Zurita J.M.Barrientos Guzman A, et al. Essential thrombocytosis with acute renal failure due to bilateral thrombosis of the renal arteries and veins. Nephron. 1982;32:73–74.
Votaw M.L.Spannuth Jr, C.Krish G, et al. Acute renal failure in a patient with essential thrombocythemia, diabetes mellitus, and heterozygous hemoglobin C disease. South Med J. 1990;83:57–59.
Farhat MH, Kuriakose P, Jawad M, et al. Sequential occurrence of thrombotic thrombocytopenic purpura, essential thrombocythemia, and idiopathic thrombocytopenic purpura in a 42- year-old African-American woman: a case report and review of the literature. J Med Case Rep. 2012;6:93.
Ardissino G, Salardi S, Colombo E, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr. 2016;175(4):465–473.
Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847–56.
Bianchi V, Robles R, Alberio L, et al. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710–3.
Mariotte E, Azoulay E, Galicier L, et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016;3:e237–45.
Reese JA, Muthurajah DS, Hovinga JAK, et al. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. Pediatr Blood Cancer. 2013;60:1676–82.
Sawler D, Parker A, Britto J, et al. Time from suspected thrombotic thrombocytopenic purpura to initiation of plasma exchange and impact on survival: a 10-year provincial retrospective cohort study. Thromb Res. 2020;193:53–9.
Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500–1.
Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med. 1991;325:393–7.
Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18:2496–502.
Terrell DR, Williams LA, Vesely SK, et al. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3:1432–6.
Medina PJ, Sipols JM, George JN. Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2001;8:286–93.
Shammas F, Meyer P, Heikkila R, et al. Thrombotic microangiopathy in a patient with chronic myelogenous leukemia on hydroxyurea. Acta Haematol. 1997;97:184–6.
Arıkan F, Yıldız Y, Ercan T, et al. Hydroxychloroquine-associated thrombotic thrombocytopenic purpura. Turk J Hematol. 2020;37:302–4.
Zuber J, Martinez F, Droz D, et al. Alpha-interferon-associated thrombotic microangiopathy: a clinicopathologic study of 8 patients and review of the literature. Medicine (Baltimore). 2002;81:321–31.
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239–45.
Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–90.
Hofmann WP, Zeuzem S. A new standard of care for the treatment of chronic HCV infection. Nat Rev Gastroenterol Hepatol. 2011;8:257–64.
O’Brien S, Kantarjian HM, Talpaz M. Practical guidelines for the management of chronic myelogenous leukemia with interferon alpha. Leuk Lymphoma. 1996;23:247–52.
Shuqin Mei, Yun Feng, Linlin Cui, et al. Thrombotic thrombocytopenic purpura developed after pegylated interferon treatment for hepatitis B infection. BMC Nephrol. 2022;23(1):400.
Kiyoshi Kitano, Yukio Gibo, Atsushi Kamijo, et al. Thrombotic thrombocytopenic purpura associated with pegylated-interferon alpha-2a by an ADAMTS13 inhibitor in a patient with chronic hepatitis C. Haematologica. 2006;91(8 Suppl):ECR34.
Lacotte L, Thierry A, Delwail V, et al. Thrombotic Thrombocytopenic Purpura during Interferon Alpha Treatment for Chronic Myelogenous Leukemia. Acta Haematol. 2000;102(3):160–2.
R Gangaraju, SJ Kim, JF Dong, et al. Thrombotic Thrombocytopenic Purpura Associated With Pegylated Interferon Alfa-2a Use in a Patient With Polycythemia Vera. J Natl Compr Cancer Netw. 2017; 15(6):757–760.
Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(12):1599–613.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654–66.
Wu WZ, Sun HC, Shen YF, et al. Interferon alpha 2a down-regulates VEGF expression through PI3 kinase and MAP kinase signaling pathways. J Cancer Res Clin Oncol. 2005;131:169–78.
Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–63.
Acknowledgements
None.
Funding
This study was supported by the Science and Technology Fund of Sichuan (2019YFS0286). The funder supported the fees upon acceptance for publication. The funders had no role in data collection, and manuscript writing or submission.
Author information
Authors and Affiliations
Contributions
CQ collected data and finished the article. DY and FL proofread and revised the manuscript. HQ reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient was informed about the availability and importance of the data, including the clinical data, images, and health information described in this article.
Consent for publication
Written informed consent was obtained from the patient for the publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare no potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qin, C., Yin, D., Liu, F. et al. Thrombotic thrombocytopenic purpura in a patient on long-term alpha-interferon therapy for essential thrombocythemia: a case report. BMC Nephrol 24, 143 (2023). https://doi.org/10.1186/s12882-023-03200-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-023-03200-7