
Abstract
Fruit length is a crucial agronomic trait of snake gourd (Trichosanthes anguina L); however, genes associated with fruit length have not been characterised. In this study, F2 snake gourd populations were generated by crossing the inbred lines, S1 and S2 (fruit lengths: 110 and 20 cm, respectively). Subsequently, bulk segregant analysis, sequencing, and fine-map** were performed on the F2 population to identify target genes. Our findings suggest that the fruit length of snake gourd is regulated by a major-effect regulatory gene. Mining of genes regulating fruit length in snake gourd to provide a basis for subsequent selection and breeding of new varieties. Genotype-phenotype association analysis was performed on the segregating F2 population comprising 6,000 plants; the results indicate that the target gene is located on Chr4 (61,846,126–61,865,087 bp, 18.9-kb interval), which only carries the annotated candidate gene, Tan0010544 (designated TFL). TFL belongs to the MADS-box family, one of the largest transcription factor families. Sequence analysis revealed a non-synonymous mutation of base C to G at position 202 in the coding sequence of TFL, resulting in the substitution of amino acid Gln to Glu at position 68 in the protein sequence. Subsequently, an InDel marker was developed to aid the marker-assisted selection of TFL. The TFL in the expression parents within the same period was analysed using quantitative real-time PCR; the TFL expression was significantly higher in short fruits than long fruits. Therefore, TFL can be a candidate gene for determining the fruit length in snake gourd. Collectively, these findings improve our understanding of the genetic components associated with fruit length in snake gourds, which could aid the development of enhanced breeding strategies for plant species.
Similar content being viewed by others


Introduction
Snake gourds (Trichosanthes anguina L; 2n = 2x = 22) are diploid, annual woody climbers of the Trichosanthes genus and Cucurbitaceae family [1]. Originating in tropical Asia [2], they are an important ornamental and edible Cucurbitaceae plant. The snake gourd fruit is rich in nutrients, including vitamins, flavonoids, carotenoids, lycopene, and phenolic acids. Ingestion of snake gourds can enhance appetite and nourishment and is beneficial for people with high blood pressure and heart disease [1, 3]. Therefore, the length of the fruit is an important ornamental horticultural trait. However, the genetic and molecular regulatory mechanisms associated with snake gourd length remain unclear, which limits the development of optimal breeding strategies for this plant.
Nevertheless, notable progress has been made in elucidating the regulatory pathways for fruit length in other members of the Cucurbitaceae family, especially in cucumbers, for which 20 quantitative trait loci (QTL) regulating fruit length and shape have been defined [4,5,6]. Weng et al. [7] mapped nine fruit size-related traits associated with cucumber using three QTL models. The MADS-box transcription factor gene, of which CsFUL1A is a functional allele, regulates fruit length along with SF1, SF2, SF3, CsCRCG, and CsaV3_1G044310. Furthermore, CsFUL1A inhibits auxin accumulation and cell division or expansion during fruit development by suppressing the expression of its downstream target, SUPERMAN (CsSUP), and auxin transporter genes, PIN-FORMED1 (CsPIN1) and CsPIN7, thus negatively regulating fruit elongation [8]. Meanwhile, the cucurbit-specific RING-type E3 ligase encoded by SF1 regulates ethylene expression to control cucumber fruit length [10]. CsaV3_1G044310 may affect type II inositol polyphosphate 5-phosphate, leading to the distribution and polar transport of auxin and thus affecting auxin-related cell expansion [11]. Small fruits are produced in response to a non-synonymous mutation in SF3—a homolog of the katanin β subunit gene, KTN1—reducing cell number [12, 13]. Meanwhile, the homolog of the Arabidopsis crab CLAW (CsCRC) gene, CsCRCG, on FS5.2 of cucumber positively regulates cell growth and fruit elongation by affecting transcriptional activation of the downstream target gene, CsARP1 [14, 15].
Additional gene functions have been reported for other members of the Cucurbitaceae family; for example, BFS on chromosome 2 in the wax gourd controls fruit shape. BFS is a member of the IQ67-domain protein family that controls Ca2+/calmodulin-dependent protein kinase II signal transduction from the cell membrane to the nucleus, thus affecting cell division and cytoskeletal rearrangement to control the fruit shape of wax gourds [16]. There are 159-bp deletions of the Cla011257 gene that belong to the IQD protein family, affecting the fruit shape in watermelons and leading to fruit elongation [17]. Therefore, in contrast to the genetic mechanisms responsible for regulating fruit shape in watermelons and wax gourds, most genes controlling fruit length in cucumbers indirectly affect the auxin-related response mechanisms of downstream target cells, effectively influencing cell number and size.
One of the largest transcription factor families in eukaryotes is the MADS-box, of which AtAGL6 is a member. AtAGL6 plays an important role in regulating flowering transformation, floral organ formation, and lateral organ development in Arabidopsis [18]. Furthermore, the MADS-box gene, FRUITFULL (FUL), downregulates the expression of IND, ALC, SHP1, and SHP2 at the valve margin, thus controlling valve elongation and fruit opening [19,20,21], resulting in fruits with reduced sizes resistant to cracking. CiMADS9 overexpression delays flowering and increases the number of leaves in Arabidopsis [1A).

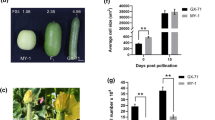
Phenotypic differences in snake gourd length. (A) Fruit length variations in S1, S2, and F1; scale bar = 10 cm. (B) Fruit length distribution in F2; scale bar = 10 cm. (C) Vertical section of commodity period fruit (18 days post-pollination) of S1 and S2; scale bar = 10 cm. (D) Cross section of commodity period fruit of S1 and S2; scale bar = 10 cm. (E) Distribution map of six generation lineages: S1 (n = 30), S2 (n = 30), F2 (n = 6,000), F1 (n = 50), BC1P1 (n = 50), BC2P2 (n = 50). (F) Microscopic images of longitudinal and transverse sections of fresh fruit from S1 and S2 18 days post-pollination; scale bar = 100 μm. Comparison of (G) longitudinal and (H) transverse sections of the average cell area and number between S1 and S2 18 days post-pollination
Cytological analysis
The mesocarp from the middle portion of the fruit was cut into thin slices and immediately placed in a formaldehyde/alcohol/acetic acid solution (50% ethanol:40% formaldehyde: glacial acetic acid = 16:1:1) in a 50-mL vial to compare the cytological properties of fruits from the two parental lines on days 9, 6, and 3 before flowering and on days 0, 6, 12, and 18 after pollination. Thin sections were paraffinised and sliced with a microtome into vertical and horizontal sections. Subsequently, the sections were deparaffinised with xylene and treated with a Safranin solid green double stain. The slides were visualised with a Z2 automatic upright differential interference fluorescence microscope (Zeiss, Germany). The cell number (X), cell area (A), and average cell area in a given section were calculated. The area of the whole-fruit longitudinal section (A′) was determined by measuring the ovary or fruit diameter and using the equation for the area of an ellipse (for a longitudinal section). The cell number in the whole-fruit cross sections or longitudinal sections (X′) was calculated using the equation X/A = X′/A′. Cell sizes and number of parental lines were quantified using Image J software (https://imagej.nih.gov/ij/https://imagej.nih.gov/ij/) and evaluated using Microsoft Office Excel 2019 (Microsoft, Redmond, WSP, USA) (Fig. 1C, D).
DNA extraction
The cetyltrimethylammonium bromide method [27] was used to extract genomic DNA from the leaves of the parent plants and the F1 and F2 populations. A k5800 ultra-micro spectrophotometer (Kaiao, Bei**g, China) was used to measure the concentration and purity of the extracted DNA, and 1.5% agarose gel electrophoresis was used to assess DNA quality.
BSA sequencing map** strategy
Of the 2,548 F2 plants created by crossing S1 and S2, 60 with extreme traits (30 long and 30 short) were selected to create two groups with notably opposing phenotypes, the phenotypes of Long and short fruit pool plants is shown in Supplementary Table S1 and S2. After filtering, two pools of extreme trait data were admixed, with one long pool and one short pool being constructed. The two mixed and two-parent pools were used for association analyses. DNA was extracted from young leaves, fragmented by ultrasonication, and subjected to fragment purification and end repair, where adenosine was added at the 3′-end, and sequencing junction ligation was performed. The DNA fragments were then separated via agarose gel electrophoresis to facilitate fragment size selection. Next, the DNA sequences were amplified using PCR to form the sequencing library. The libraries were then sequenced using the Illumina sequencing platform, and raw image data files obtained were transformed into Sequenced Reads after Base Calling analysis. The raw reads were filtered to obtain clean reads, which were used for subsequent analysis. The main data filtering steps included (1) removing reads with adapters; (2) filtering reads with an N content > 10%; (3) removing reads with a mass value < 10 bases and > 50%. Sequencing reads were compared with the reference genome using BWA software and located to the reference genome for subsequent mutation analysis.
Single-nucleotide polymorphisms (SNPs) and InDels were detected using GATK software. Based on the results of the positioning of clean reads in the reference genome, GATK was used for local realisation and other pre-treatments to ensure the accuracy of SNP detection and for SNP detection to determine the SNP site. The filtering criteria were as follows: SNP loci with multiple (> 2) genotypes, SNP and InDel loci with read support < 5, no polymorphism between parents, and loci missing from a sample. The Euclidean distance (ED) association and Δ SNP-index algorithms were used as association analysis techniques [28, 29].
Fine-map**
Based on the BSA-seq data and the distribution and density of the physical InDel locations, we designed seven InDel markers for each 1-Mb interval to further narrow the candidate gene regions. The primers used in this study were designed using Primer Premier 5 (Premier, Canada). Each 12-µL reaction volume contained 2 µL DNA template, 1 µL of each forward and reverse primer (10 μm), 5 µL Master Mix, and 3 µL ddH2O. The PCR cycle conditions were as follows: 95 °C for 5 min, 30–35 cycles of 95 °C for 30 s, 50–58 °C for 30 s, 72 °C for 30 s, extension at 72 °C for 5 min, and incubation at 4 °C. The digested and PCR amplification products were separated for 1 h at 300 V on a native 8% polyacrylamide gel. The resulting polyacrylamide gel is presented in Supplementary Table S3. A total of 526 long-fruit and 493 short-fruit plants from 2,548 F2 individuals were used for genotype-phenotype analysis. To further narrow down the map** range, five new InDel markers were applied to genotype the F2 population (n = 6,000 individuals) and identify recombinants. The genotypes of the recombinant plants and the most likely target gene region were inferred using a genotype-phenotype joint analysis.The primer sequence is shown in Supplementary Table S4.
Cloning and sequencing analysis of candidate genes
Forward and reverse primers were designed according to the coding sequence (CDS) of the gene (Supplementary Table S4). Tender leaf RNA was extracted, reverse transcribed into cDNA, and amplified with 2× Phanta Max Master Mix (Vazyme, Nan**g, China). Axyprep DNA gel extraction kits (Axygen, Union City, CA, USA) were used for the recovery and purification of target bands from agarose gels. For subsequent experiments, pure DNA extracted from the gels was used.
A Zero Blunt TOPO PCR Cloning Kit (CV16; Aidlab, Bei**g, China) was used to construct the recombinant plasmid, which was then cloned in Escherichia coli DH5α competent cells. Subsequently, colony PCR and agarose gel electrophoresis was performed to detect positive bacterial colonies; the five bacterial colonies with the brightest DNA bands were sent to Shenggong Biotechnology Co. (Shanghai, China) for sequencing. DNAMAN V.9 (Lynnon Biosoft, CA, United States) was used to compare amino acid and DNA sequences (Supplementary Table S5).
Analysis of spatiotemporal gene expression
To analyse gene expression in different parts and stages, RNA was extracted from the parent root, stem, and leaf. RNA from the ovary during different development stages (9, 6, and 3 days before flowering and 0, 6, 12, and 18 days after pollination) was extracted using an Eastep Super Total RNA extraction kit (Promega, Shanghai, China) following the manufacturer’s instructions. The primers for histone H3 (Tan0019208.1) and other candidate genes were designed using Primer Premier 5.0. The cDNA was synthesised using reverse transcriptase RT Master Mix (RR036A) following the manufacturer’s instructions (TaKaRa, Bei**g, China). Each quantitative reverse transcription PCR (qRT-PCR) mixture was prepared and the quantitative fluorescence was analyzed according to the methods described by Huo et al. [30]. Each quantitative reverse transcription PCR (qRT-PCR) mixture contained 2 µL cDNA, 0.8 µL forward and reverse primers, 10 µL TB Green Premix Ex TaqII, 0.4 µL ROX Reference Dye II, and 6 µL nuclease-free water. Quantitative fluorescence analysis was performed using the Applied Biosystems 7500 real-time PCR instrument (Foster City, CA, USA). Briefly, the samples were preheated at 95 °C for 30 s, followed by heating for 5 s at 95 °C, and 40 cycles at 60 °C for 34 s. High-resolution melting was performed at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. Each sample was tested for relative gene expression using the 2−∆∆Ct method for three biological replicates [31] subsequently analyzed using SPSS 25.0.
Phylogenetic analyses
To understand the relationship between the TFL protein sequence and other homologous sequences, the gene sequence was downloaded in FASTA format from the NCBI database and performed an NCBI BLAST search (NCBI, Bethesda, MD, USA). Subsequently, using the MEGA-X software’s neighbour-joining function, we created a phylogenetic tree with 1,000 replicates (Supplementary Table S6).
Molecular marker-assisted selection test
Based on the InDel differences of TFL in the parents, located in the intron, InDel markers were developed according to candidate gene sequences using Primer Premier 5 software. Bei**g Tsingke Biotechnology Co., Ltd. (Nanning, China) synthesised all primers used in this study (Supplementary Table S4). The same steps were followed as those in Sect. 2.5 for the InDel marker generation. Genotype-phenotype analysis was performed for all seed resources tested. Twenty-seven germplasm resources of snake gourd were validated and divided into long-fruited and short-fruited groups (Supplementary Table S7).
Results
Phenotypic and genetic characteristics of fruit length
Longitudinal fruit diameter (fruit stalk to receptacle) in the parents was measured 9, 6, and 3 days before flowering and 0, 6, 12, and 18 days after pollination until fruit length was set (Fig. 2R), and the growth curve was plotted (Fig. 2O). BC1P1 fruit lengths ranged from 38.4 to 85.0 cm with a mean of 55.9 cm and a standard deviation of 12.6. BC1P2 fruit lengths ranged from 15.0 to 56.0 cm with a mean of 31.2 cm and a standard deviation of 7.5. The rapid-growth period of the snake gourd occurred 6–18 days after pollination; the fruit length in the F2 population conformed to a normal distribution (Fig. 1B, E). Fruit lengths of < 30 cm and > 50 cm were classified as short and long, respectively. Of the 6,000 F2 plants, 1,189 had short-fruit, and 1,269 had long-fruit. No differences in cell proliferation rate or cell size were observed between S1 and S2 before pollination. Mostly intermediate, with a relatively small number of extremely long and extremely short fruits. However, differences arose after pollination, with S1 having a much higher rate of cell proliferation than S2. Meanwhile, the cell area amplification rate was markedly higher in S2 than in S1. On day 18, S1 had seven times more cells than S2, and S2 had twice the cell area of S1. Although the number of cells decreased after 12 days of pollination, the increase in cell area compensated for the quantity. At fruit maturity, the length of S1 fruit was five times greater than S2 (Figs. 1F-H and 2A-N, P and Q).

(A)–(G) Longitudinal section of the ovary in the S1 inbred line on days 9, 6, and 3 before flowering and on days 0, 6, 12, and 18 after pollination (scale bar = 100 μm). (H)–(N) Longitudinal section of the ovary in the S2 inbred line on days 9, 6, and 3 before flowering and days 0, 6, 12, and 18 after pollination. (O) Fruit length growth cycle curve of S1 (green) and S2 (orange). (P) Average cell size during fruit development in S1 and S2. (Q) Cell proliferation during fruit development in S1 and S2. (R) From left to right: days 9, 6, and 3 before flowering and day 0 after pollination (scale bar = 1 cm); days 6, 12, and 18 after pollination (scale bar = 10 cm)
Fine-map** and identification of the candidate gene
-
(a)
Whole genome sequencing of bulks and parental lines.
After whole-genome resequencing, 238,457,730, 221,257,766, 234,769,954 and 224,787,160 of Reads numbe were obtained from the S1, S2 ,and two extremes of mixed pools populations, respectively. The Q30 and GC values for S1 are 94% and 38%, respectively, with a size of 35.7 GB, while the corresponding values for S2 are 93% and 37%, respectively, with a size of 33.1 GB.The corresponding values for the two extremes of the mixed pool are 93% and 38%, 92% and 38%, respectively, with sizes of 35.2 GB and 33.7 GB. The statistics of sequencing data is in the Supplementary Table 8. Compared with the reference genome, the similarity rate between parents and the F2 population was 91.09–91.58%. The average sequencing depth of the parental genome was 33.6×.
-
(b)
QTL identification for fruit length.
Before association analysis, SNPs and InDels were filtered, resulting in the identification of 1,343,961 SNPs and InDel markers. We used the indexing algorithm to analyse the association between SNP markers and selected the region with a threshold > 1.35 and the Δ SNP-index values is 99%. Candidate intervals were calculated using the ED and SNP-index algorithms. Because the SNP-index interval was within the ED algorithm interval, we selected the concatenation interval (59.677,903 bp − 63,802,574 bp, Δ SNP-index value is 0.71).
-
(c)
QTL validation using single marker analysis.
Developed one marker per 1-M interval within the interval. Seven InDel markers were first developed with 526 long-fruit and 493 short-fruit plants from 2,548 F2 plants, and a genotype-phenotype association analysis was performed to compress the interval between the SG60 (60,655,536 bp) and SG62 (63,389,272 bp) markers. Five new InDel markers were then developed within this de-interval, the genotype-phenotype of 1,269 long-fruit and 1,189 short-fruit plants from 6000 F2 plants were analyzed, and five recombinant plants were identified in the extreme phenotype plants. ID is F2-19-1, F2-25-76, F2-5-16,F2-21-76 and F2-27-65 (Supplementary Table S9). As shown in Fig. 3A-C, the genotypes of F2-19-1 and F2-25-76 are shown as short fruits in marker SG61.7, and the genotype of F2-5-16 is shown as short fruits in marker SG61.8, and all the three fruit lengths are short fruits. The genotypes of F2-21-76 is shown as long fruits in marker SG61.7, and the genotype of F2-27-65 is shown as long fruits in marker SG61.8, aand all the two fruit lengths are long fruits. Therefore, the interval was narrowed down to SG61.7 (61,846,126 bp) - SG61.8 (61,865,087 bp), a total of 18.9 kb, by genotype-phenotype association analysis, and only one gene, Tan0010544 (TFL), was found in this region. Therefore, TFL was a candidate gene for controlling the fruit length of snake gourds (Fig. 3A–C).

Genetic map** of the gene regulating fruit length in snake gourds. (A) Distribution of the Euclidean distance (ED) association values on chromosomes (chr). The abscissa indicates the chr name, the coloured dots represent the ED value of each single nucleotide polymorphism (SNP) locus, the black line represents the fitted ED value, and the red dotted line represents the significance association threshold. The higher the ED value, the better the association effect of the SNP locus. (B) Fine-map** of the fruit length gene using InDels. Using 2,548 F2 individuals resulting from the S1 × S2 cross, the location of the gene regulating fruit length was narrowed down to a 2.7-M region between the markers, SG60 and SG62, on chr 4. (C) Genoty** of recombinant plants from 6,000 F2 individuals resulting from the S1 × S2 cross. The fruit length of the two parents and their F1 offspring and the genotype of the recombinant single parent are shown on the right. The location of the gene that regulates the length of the fruit was narrowed down to an 18-kb region. (D)Tan0010544 (TFL) contains the c.202 C > G mutation in the coding sequence and p.Gln68Glu substitution in the protein sequence. (E) Alignment of Tan0010544 (TFL) homologs from different species. The E68Q mutation in S1 is highlighted in red
-
(d)
Candidate gene identification.
Candidate gene and their whole CDSs were cloned. To determine the TFL sequence, we cloned its CDS from the two parental lines and performed a sequence comparison analysis. The total lengths of TFL and its CDS were 11.02 kb and 741 bp, respectively. Sequence analysis revealed a non-synonymous mutation of base C to G at position 202 in the coding sequence of TFL, resulting in a substitution of amino acid Gln to Glu at position 68 in the protein sequence (Fig. 3D). Alignment of the S1 and S2 amino acid sequences with those of homologous proteins from various plant species showed that E–68 is conserved among land plants (Fig. 3E). The TFL determines the interval inSupplementary Table 10.
Gene expression analysis
The expression of TFL was compared between the root, stem, leaf, and male flower in S1 and S2. Furthermore, TFL expression was examined at different periods during ovary formation, particularly on days 9, 6, and 3 before flowering and days 0, 6, 12, and 18 after pollination. qRT-PCR analysis revealed no difference in TFL expression between roots, leaves, and ovary 6 days after pollination. However, significant differences (0.01 < P < 0.05) were observed in the ovary 6 days before flowering and 0 days after pollination. The expression of TFL in the stem and male flower differed significantly (P < 0.01) on day 9 before flowering and on days 12 and 18 after pollination. Comprehensive analysis of paraffin sections and the growth cycle revealed an inverse correlation between cell number and area, with higher TFL expression levels corresponding to shorter fruit length and reduced cell numbers, yet larger cell areas (Figs. 2A–O and 4). The expression of TFL increased significantly in parents 12 days after pollination, and this led us to suspect that this gene may be related to cell development (Figs. 2P–Q and 4). Meanwhile, the regulatory mechanisms linking TFL with fruit length remain unknown.

Quantitative real-time PCR analysis to assess the relative expression of Tan0010544 (TFL) in roots, stems, leave and male flowers of the S1 and S2 inbred lines and at different periods during the ovarian formation, particularly on days 9, 6, and 3 before flowering and days 0, 6, 12, and 18 after pollination using quantitative real-time PCR. *P < 0.05, **P < 0.01
Phylogenetic analysis
The results of the phylogenetic analysis revealed that TFL had a close phylogenetic relationship with legumes and Cucurbitaceae, indicating that TFL was evolutionarily conserved in Cucurbitaceae. Sequence alignment results indicated that TFL shares 95% sequence homology with XP_022998764.1 found in Cucurbita maxima. (Fig. 5).

Phylogenetic tree of Tan0010544 (TFL) and its homologous proteins. The phylogenetic tree was constructed using MEGA-X software with 1,000 bootstrap replications. The numbers at the tree nodes indicate bootstrap values
Development of InDel markers for molecular marker-assisted breeding
In total, 27 snake gourd inbred lines with extreme fruit length differences, including 9 long and 18 short fruits, were selected for validation using the InDel molecular marker. We observed that the bands of 9 long-fruited snake gourd materials were consistent with S1, whereas those of 16 short-fruited snake gourds were consistent with S2. However, the bands from samples 26 and 27 were inconsistent with those of F1. The correlation rate between genotype and phenotype was 92% (Fig. 6). Collectively, our results suggest that the InDel marker can be used in the molecular marker-assisted breeding of snake gourd fruit length.

In total, 27 different lines of snake gourd were analysed using InDel markers. The first 9 and last 16 corresponded to long and short fruit parent phenotypes, respectively. We observed inconsistencies between the genotypes and phenotypes of species 26 and 27 in relation to those of their parents
Discussion
Relationship between fruit length and TFL expression in snake gourds
In this study, we used a pair of inbred snake gourd lines with significant differences in fruit lengths as parental strains. A major effect gene affecting the fruit length of the snake gourd was located using the BSA-seq method. Extreme long and extreme short fruits were used for primary localisation, similar in fruit length to the long-fruited parent and short-fruited parent, respectively, to obtain the BSA primary interval. The results showed that the interval was 59,677,903 bp − 63,802,574 bp on chromosome 4. The intervals exceeding 99% of the Δ SNP-index value were selected as the intervals to be finely localised later, and for the sake of the accuracy of the experiment, I discarded the intermediate values of the F2 monocots, and only the phenotypically extreme monocots were used for the fine localisation. We narrowed the interval using extreme phenotypes to 18 kb according to the genotype-phenotype approach and confirmed that Tan0010544 (TFL), a MADS-box family gene, implicates that TFL is involved in regulation of fruit length. We also confirmed by qRT-PCR analysis that TFL expression does not differ significantly between roots and leaves but is significantly different in stems and flowers. Although the lengths of the ovaries and the cell number and area of the parents 9 days before flowering did not differ significantly, TFL expression was markedly different. The expression of TFL at 9 days before flowering coincides with the differential growth rate of the ovary, leading to a two-fold difference in fruit length at flowering. The fruit length of S2 0 days after pollination was half that of S1. The ovaries underwent rapid growth and development 6 days after pollination. Starting from 9 days pre-pollination to 18 days after pollination, TFL expression increased significantly; this was mirrored, subsequently, by a significant change in fruit length. By 12 days post-pollination, the S1 and S2 fruit length increased five times and 2.5 times, respectively, compared to 0 days after pollination. Additionally, TFL expression in S1 increased significantly, whereas the growth rate and rate of fruit length growth decreased.
Collectively, the growth cycle curve and gene expression in parents suggest that the Tan0010544 (TFL) gene is associated with the fruit length of snake gourds. We speculate that the TFL gene may affect the auxin regulation pathway to regulate fruit growth in snake gourds. Auxin plays a crucial role in cell division and differentiation, as well as in fruit development [32]. Therefore, TFL may also affect cell number and area. Although the length of the parents was the same 9 days before flowering, the difference in TFL expression corresponded with cell division and differentiation. The paraffin sections did not reveal significant differences in the number or area of S1 and S2 cells 9 days before flowering, whereas the area of S2 cells was two times greater than that of S1 during the last 18 days. Meanwhile, there were seven times more S1 cells than S2 cells. Collectively, the increase in the number of cells in S1 than in S2 may contribute to the increase in the fruit length of S1. Thus, TFL could affect cell division and regulate the fruit length of snake gourds.
Association of TFL function with the MADS-box transcription factor family
The MADS-box gene contains a highly conserved region that encodes a MADS-box domain composed of approximately 60 amino acids. Previous phylogenetic analysis of the MADS-box gene in fungi, plants, and animals [33] has revealed that the gene underwent a replication event before differentiation between animals and plants, producing type I (SRF-like) and type II (MEF2-like) lineages. Using the structure of A. thaliana for duplication and motif analysis, type I and type II MADS-box genes were further divided into five subtypes: Mα, Mβ, Mγ, Mδ, and MIKC [34]. Phylogenetic analysis of the rice MADS-box gene family revealed that the type I gene contains Mα, Mβ, Mγ, and Mδ subfamilies, whereas type II comprises MIKC subgroups [35]. The MADS-box gene family reportedly encodes many transcription factors that play a vital role in the growth and development of various plants, including fruit growth and development [21], seed colour change, and embryo development [36]. It also contributes to stress resistance, as to well as cell and organ differentiation. There are several reports on the MADS-box family that affect flower development. For example, APETALA1, APETALA3, and PISTILLATA act as bifunctional transcription factors (activating and inhibiting factors) and affect flower patterning and fruit ripening [37,38,39]. Zhao et al. [8] reported that the MADS-box family gene, CsFUL1A, affects fruit length by regulating auxin, cell number, and size.
In this study, the expression of TFL in the flowers and stems of the parents also differed significantly. The MADS-box family affects the growth and development of flowers, and TFL expression was reportedly significantly different in floral organs. Smaczniak et al. [40] reported that the MADS-box transcription factor can bind to the CArG-box of the target gene. CsSUP has three CArG boxes in the promoter region, one CArG-box in the coding region, and one type A CArG-box located 2,200 bp from the transcription start site and binds to the MADS-box protein [41]. Zhao et al. [8] confirmed that CsFUL1A binds to the CArG-box of the promoter region, inhibiting the expression of SUPERMAN, a regulator of cell division and expansion. Collectively, we speculate that the TFL gene may also combine with the CArG-box region of the SUPERMAN promoter and subsequently inhibit the expression of SUPERMAN to further inhibit cell division and expansion. This could inhibit the expression of the auxin transporters, PIN-FORMED1 (PIN1) and PIN7, both of which jointly control the fruit length of snake gourds, reducing the accumulation of auxin. TFL encodes a typical type II MADS-box protein with four modular domains (MIKC), the N-terminal DNA-binding MADS domain (M), the intervening (I) and keratin-like (K) regions, and a C-terminal region (C) (Supplementary Table S11 and Supplementary Fig. 1).
In this study, a major-effect gene (TFL) regulating fruit length was cloned for the first time in a pair of snake gourds with significant differences in fruit length using the BSA method combined with genotype-phenotype analysis of F2 recombinant plants. Furthermore, a functional molecular marker was developed for molecular marker-assisted selection in snake gourds. Samples 26 and 27 may be affected by other non-alleles, and these two materials also provide a material basis for us to continue to explore other non-alleles of snake gourd fruit length, and we will also carry out the study of non-alleles of snake gourd fruit length in these two materials, so as to provide a theoretical basis for comprehensively analyzing the basis of the molecular regulation of snake gourd fruit length. To our knowledge, this is the first report of a gene related to fruit length in snake gourds. This study provides novel insights into snake gourd rapid breeding and could help improve the efficient selection of new varieties of snake gourd for fruit length orientation. In China, snake gourd is mostly used as an ornamental crop, attracting consumers with its exaggerated fruit length. Moreover, our data provide a theoretical basis for future analyses focused on delineating the underlying mechanisms that regulate fruit length at the genetic level. This may allow for a more comprehensive domestication of the Cucurbitaceae family, and a more controlled manipulation of their traits for agricultural applications.
TFL belongs to the MADS-box family, one of the largest transcription factor families. Sequence analysis revealed a non-synonymous mutation of base C to G at position 202 in the coding sequence of TFL, resulting in the substitution of amino acid Gln to Glu at position 68 in the protein sequence. We speculate that the TFL gene may affect the auxin regulation pathway to regulate fruit growth in snake gourds. An InDel marker was developed to aid the marker-assisted selection of TFL.
Data availability
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0004134. https://db.cngb.org/search/?q=CNP0004134. The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
Devi N. Medicinal values of Trichosanthus Cucumerina L. (snake gourd) - a review. Br J Pharm Res. 2017;16:1–10.
Adebooye OC. Phyto-constituents and anti-oxidant activity of the pulp of snake tomato (Trichosanthes cucumerina L). Afr J Tradit Complement Altern Med. 2008;5:173–9.
Yusuf AA, Folarin OM, Bamiro FO. Chemical composition and functional properties of snake gourd (Trichosanthes cucumerina) seed flour. Niger Food J. 2007;25:36–45.
Che G, Zhang X. Molecular basis of cucumber fruit domestication. Curr Opin Plant Biol. 2019;47:38–46.
Pan Y, Wang Y, McGregor C, Liu S, Luan F, Gao M, Weng Y. Genetic architecture of fruit size and shape variation in cucurbits: a comparative perspective. Theor Appl Genet. 2020;133:1–21.
Sheng Y, Pan Y, Li Y, Yang L, Weng Y. Quantitative trait loci for fruit size and flowering time-related traits under domestication and diversifying selection in cucumber (Cucumis sativus). Plant Breed. 2020;139:176–91.
Weng YQ, Colle M, Wang YH, Yang LM, Rubinstein M, Sherman A, et al. QTL map** in multiple populations and development stages reveals dynamic quantitative trait loci for fruit size in cucumbers of different market classes. Theor Appl Genet. 2015;128:1747–63.
Zhao JY, Jiang L, Che G, Pan YP, Li YQ, Hou Y, et al. A functional allele of CsFUL1 regulates fruit length through repressing CsSUP and inhibiting auxin transport in cucumber. Plant Cell. 2019;31:1289–307.
**n TX, Zhang Z, Li S, Zhang S, Li Q, Zhang ZH, et al. Genetic regulation of ethylene dosage for cucumber fruit elongation. Plant Cell. 2019;31:1063–76.
Zhang Z, Wang B, Wang S, Lin T, Yang L, Zhao Z, et al. Genome-wide target map** shows histone deacetylase complex1 regulates cell proliferation in cucumber fruit. Plant Physiol. 2020;182:167–84.
Cheng F, Song M, Cao L, Zhang M, Yang Z, Chen J, et al. Genetic map** for a medium short-fruit mutant of cucumber. Acta Hortic. 2021;48:1–12.
Wang H, Sun J, Yang F, Weng Y, Chen P, Du S, et al. CsKTN1 for a katanin p60 subunit is associated with the regulation of fruit elongation in cucumber (Cucumis sativus L). Theor Appl Genet. 2021;134:2429–41.
Song M, Fu W, Wang Y, Cheng F, Zhang M, Chen J, Lou Q. A mutation in CsKTN1 for the Katanin p60 protein results in miniature plant in cucumber, Cucumis sativus L. Veg Res V. 2022;2:1–9.
Che G, Pan Y, Liu X, Li M, Zhao J, Yan S, et al. Natural variation in CRABS CLAW contributes to fruit length divergence in cucumber. Plant Cell. 2023;35:738–55.
Pan Y, Chen B, Qiao L, Chen F, Zhao J, Cheng Z, Weng Y. Phenotypic characterization and fine map** of a major-effect fruit shape QTL FS5.2 in cucumber, Cucumis sativus L., with near-isogenic line-derived segregating populations. Int J Mol Sci. 2022;23:13384.
Cheng Z, Liu Z, Xu Y, Ma L, Chen J, Gou J, et al. Fine map** and identification of the candidate gene BFS for fruit shape in wax gourd (Benincasa hispida). Theor Appl Genet. 2021;134:3983–95.
Dou JL, Zhao SJ, Lu XQ, He N, Zhang L, Ali A, et al. Genetic map** reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L). Theor Appl Genet. 2018;131:947–58.
Yoo SK, Wu X, Lee JS, Ahn JH. Agamous-like 6 is a floral promoter that negatively regulates the FLC/MAF clade genes and positively regulates FT in Arabidopsis. Plant J. 2011;65:62–76.
Gu Q, Ferrándiz C, Yanofsky MF, Martienssen R. The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development. 1998;125:1509–17.
Ferrándiz C, Liljegren SJ, Yanofsky MF. Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science. 2000;289:436–8.
Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, Yanofsky MF. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature. 2000;404:766–70.
Zhang JY, Mo ZH, Li YR, Gang W, Ji-** X, **ao-Dong J, et al. Cloning and functional analysis of the MADS-box CiMADS9 gene from Carya illinoinensis. Hortic Plant J. 2015;1:11–6.
Zhang H, Forde BG. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science. 1998;279:407–9.
Mena M, Mandel MA, Lerner DR, Yanofsky MF, Schmidt RJ. A characterization of the MADS-box gene family in maize. Plant J. 1995;8:845–54.
Thompson BE, Bartling L, Whipple C, Hall DH, Sakai H, Schmidt R, Hake S. Bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell. 2009;21:2578–90.
Ma LL, Wang Q, Mu JL, Fu AZ, Wen CL, Zhao XY, et al. The genome and transcriptome analysis of snake gourd provide insights into its evolution and fruit development and ripening. Hortic Res. 2020;7:199.
Porebski S, Bailey LG, Baum BR. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep. 1997;15:8–15.
Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ. MMAPPR: mutation map** analysis pipeline for pooled RNA-seq. Genome Res. 2013;23:687–97.
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, et al. MutMap+: genetic map** and mutant identification without crossing in rice. PLoS ONE. 2013;8:e68529.
Huo Y, Zhang G, Yu W, Liu Z, Shen M, Zhao R, et al. Forward genetic studies reveal LsAPRR2 as a key gene in regulating the green color of pericarp in bottle gourd (Lagenaria siceraria). Front Plant Sci. 2023;14:1130669.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆Ct method. Methods. 2001;25:402–8.
Zhao Y. Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol. 2010;61:49–64.
Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A. 2000;97:5328–33.
Parenicová L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15:1538–51.
Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, Kapoor S. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007;8:242.
Nesi N, Debeaujon I, Jond C, Stewart AJ, Jenkins GI, Caboche M, Lepiniec L. The TRANSPARENT TESTA16 locus encodes the Arabidopsis BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell. 2002;14:2463–79.
Kaufmann K, Wellmer F, Muiño JM, Ferrier T, Wuest SE, Kumar V, et al. Orchestration of floral initiation by APETALA1. Science. 2010;328:85–9.
Smaczniak C, Immink RGH, Muiño JM, Blanvillain R, Busscher M, Busscher-Lange J, et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc Natl Acad Sci U S A. 2012b;109:1560–5.
Wuest SE, O’Maoileidigh DS, Rae L, Kwasniewska K, Raganelli A, Hanczaryk K, et al. Molecular basis for the specification of floral organs by APETALA3 and PISTILLATA. Proc Natl Acad Sci U S A. 2012;109:13452–7.
Smaczniak C, Immink RG, Angenent GC, Kaufmann K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development. 2012a;139:3081–98.
Riechmann JL, Krizek BA, Meyerowitz EM. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc Natl Acad Sci U S A. 1996;93:4793–8.
Funding
This work was supported by the Guangxi Science and Technology Major Special Project (GuiKe AA17204039-2 and GuiKe AA22068088-2).
Author information
Authors and Affiliations
Contributions
WY and PW conceived and designed the experiments. QJ and YW performed the experiments. SH, JZ, YL, HF, BZ, and CZ collected the data. QJ and YX analysed the data. QJ wrote the manuscript. WY, PW, and YX revised the manuscript. QJ, PW, and YX contributed equally. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, Q., Wang, P., Xu, Y. et al. Fine map** of TFL, a major gene regulating fruit length in snake gourd (Trichosanthes anguina L). BMC Plant Biol 24, 286 (2024). https://doi.org/10.1186/s12870-024-04952-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-04952-6