
Abstract
Background
Mountains of the world host a significant portion of all terrestrial biodiversity, and the region of the Qinghai-Tibet Plateau (QTP) stands as one of the most remarkable mountain regions on Earth. Because many explosive radiations occurred there, the QTP is a natural laboratory which is ideal to investigate patterns and processes linked to speciation and diversification. Indeed, understanding how closely related and sympatric species diverged is vital to explore drivers fostering speciation, a topic only rarely investigated in the QTP. By combining genomic and environmental data, we explored the speciation process among three closely related and sympatric species, Gentiana hexaphylla, G. lawrencei and G. veitchiorum in the QTP region.
Results
Combining genome sizes and cytological data, our results showed that G. hexaphylla and G. veitchiorum are diploid, whereas G. lawrencei is tetraploid. Genetic clustering and phylogenetic reconstruction based on genomic SNPs indicated a clear divergence among the three species. Bayesian clustering, migrant, and D-statistic analyses all showed an obvious signature of hybridization among the three species, in particular between G. lawrencei and both G. hexaphylla and G. veitchiorum in almost all populations. Environmental variables related to precipitation and particularly temperature showed significant differences among the three gentians, and in fact a redundancy analysis confirmed that temperature and precipitation were the major climatic factors explaining the genetic differentiation among the three species.
Conclusion
Our study suggested that ancient hybridization, polyploidization, geological isolation and the evolution of different climatic preferences were all likely to be involved in the divergence of the three Gentiana species, as may be the case for many other taxa in the QTP region.
Similar content being viewed by others

Introduction
Several major mountain systems of the world are remarkable centres of species diversity, and many of them are classified as hotpots of biodiversity [1,2,3,4]. The accumulation of biodiversity in mountains, over evolutionary times, is usually associated with geological processes (e.g., uplift, erosion) and climatic changes (e.g., climate fluctuations) because they combine to generate a complex topography on which a plethora of highly heterogeneous environments co-exist along altitudinal gradients [2, 5, 6]. This heterogeneity fosters for example divergent natural selection and adaptive radiation [7, 8], which were shown to partly explain the uneven distribution of biodiversity in species richness on Earth [9, 10]. Furthermore, diversification may be promoted by climate-driven cycles modifications of distribution ranges, via a so-called species pump effect [3]. Indeed, depending on the climate state (e.g., Last Inter Glacial (LIG) or Last Glacial Maximum (LGM)), mountain ranges may act either as barriers or facilitators of dispersal, causing distribution ranges of organisms to alternate between fragmentation (isolation; allopatric speciation) [2, 11] and expansion (leading to secondary contact; hybridization) [12, 13].
The highest and largest mountain region of the world, the Qinghai-Tibet Plateau (QTP) region, which includes the QTP platform, the Himalayas and particularly the Hengduan Mountains (HM) [14], harbours a rich and probably old alpine flora [23], hinting at a parallel effect of habitat heterogeneity on the evolution of reproductive isolation and speciation. Furthermore, a few studies pointed out the crucial role of hybridization in sha** the flora of the QTP region [24], but overall, its extent is probably vastly underestimated there [25]. For example, in the genus Saxifraga, the occurrence of hybridization has been reported numerous times in all European clades, whereas it remains almost undetected in the QTP region [26] despite the much larger number of species in the latter area.
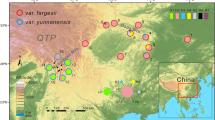
The alpine biome of the QTP region is the centre of diversity and the biogeographical origin of many species-rich taxa, including Gentiana (Gentianaceae) which is distributed in almost all temperate areas of the world [27, 28]. Gentiana is composed of 13 sections [29], some of which being endemic or near endemic to the region of the QTP. This is for example the case of G. section Kudoa (Masam.) Satake & Toyok. ex Toyok with 25 out of 26 species occurring there [27, 29]. Section Kudoa continuously attracted biologists’ attention due to its horticultural and medicinal value [27], its taxonomic complexity [29] and its relevance for evolutionary studies [30]. Previous phylogenetic studies showed a relatively rapid diversification of section Kudoa since the Pliocene [30, 31]. Although ploidy levels are not known for all species in this section, five of them are known to be diploids and two are tetraploids [32,33,34,35]. In this study, we focused on three closely related species in G. section Kudoa, namely G. hexaphylla Maxim., G. veitchiorum Hemsl. and G. lawrencei Burkill of which distribution ranges are largely sympatric (Fig. 1)[27]. The three gentians are used in traditional Chinese and Tibetan Medicine and domesticated for horticultural use. The three species can be distinguished by morphological traits such as the shape and arrangement of their leaves (e.g., opposite or in whorls), and the color and shape of their corolla [36]. All three species are perennials and characterized by little pre-zygotic isolation with most visitations being from generalists such as bumblebees [37; personal observations]. Spatial genetic structures were investigated in all three species, unveiling a north-western and south-eastern clade in both G. veitchiorum and G. lawrencei [19], as well as a northern and southern clade in G. hexaphylla [38]. These spatial genetic structures at least partly derive from a combination of climate-driven range displacement and geological barriers [19, 38], but it remains unclear whether their respective environmental preferences also contributed to the genetic patterns observed. In addition, at least one clade of G. lawrencei is likely to have experienced hybridization with G. veitchiorum in one refugium shared between the two species [19]. Indeed, as in Saxifraga, hybridization may be more common than previously thought in the QTP region for Gentiana [39,40,41] given the number of cases of hybridizationdetected in Europe [e.g., 25, 42, 43] and the much larger number of closely-related species in the QTP region [27].
Here, to better understand the mechanisms at work in the process of speciation in Gentiana, we combined genomic and climatic data to detect the factors that may have contributed to the divergence among G. hexaphylla, G. veitchiorum and G. lawrencei by sampling across known and spatially structured populations. We specifically aimed to answer (1) Did geographical isolation and climate preferences foster differentiation among the three Gentiana species? (2) How did hybridization affect their divergence?
Results
Genome size
For genome size estimation, four replications were performed for G. hexaphylla and G. veitchiorum, and three for G. lawrencei. The mean values of the genome size of G. hexaphylla, G. veitchiorum and G. lawrencei were 3.18 G, 3.25 G and 5.00 G, respectively. The standard deviation in the three species ranged from 0.058 to 0.100 (Table S1).
Data preprocessing and SNP calling
Individuals of G. veitchiorum and G. lawrencei were newly sequenced in this study, and the raw data of 35 individuals of G. hexaphylla were retrieved from Fu et al. [38]. After quality filtering, the number of reads retained per sample varied from 4.45 × 106 to 3.18 × 107, with a median value of 1.04 × 107 (Table S2). The depth per sample varied from 5.76 × to 26.97 ×, averaged at 12.53 ×. After filtering for MAF, linkage-disequilibrium (LD) and missing data, the total number of unlinked SNPs obtained for all samples was 143,611. When the outgroup was included, 144,402 SNPs were kept for downstream analysis.
Population genetic structure and genetic divergence
Genomic SNPs showed that G. lawrencei had a slightly higher genetic diversity (e.g., Ar, Ho) than G. hexaphylla and G. veitchiorum (Table 1). The mean Ar vs. Ho in G. lawrencei, G. hexaphylla and G. veitchiorum were 1.1839 vs. 0.1626, 1.1633 vs. 0.1545 and 1.1542 vs. 0.1228, respectively. The Mantel test showed no-significant negetive correlations between altitude and Ar (r2 = 0.042, p = 0.402) and Hoo (r2 = 0.042, p = 0.399), respectively. The CV errors from Admixture analyses showed lowest value at K = 3 (Fig. S1), indicating the three species shall be clustering into three groups, so the inferred three genetic clusters corresponded to the three species included in this study (Fig. 2B). Based on the clustering analyses, introgression was detected from G. lawrencei to G. hexaphylla and G. veitchiorum, and from G. veitchiorum to G. lawrencei. One individual in a population (Fu2016087) of G. hexaphylla contained almost equal genetic composition of G. hexaphylla and G. lawrencei, respectively. From the PCA plot, the first principal component (PC1), which explained 20.46% of all genetic variance, differentiated the three species; the second principal component (PC2), which explained 13.25% of all genetic variance, differentiated the three species as well (Fig. 2 A). Generally, the pairwise FST values were much higher among species than within species (Fig. 3 A). Detailed pairwise FST values between populations were showed in Table S3. The weighted FST values between G. hexaphylla and G. veitchiorum, G. hexaphylla and G. lawrencei, and G. veitchiorum and G. lawrencei were 0.237, 0.187, 0.149, respectively. Plotting FST/(1-FST) and geographic distances among populations in the three Gentiana species showed a significant positive correlation in both within species (r2 = 0.624, p < 2.2e− 16) and between species (r2 = 0.089, p = 0.0007) (Fig. 3B).

Distribution ranges of three Gentiana species based on locality data retrieved from GBIF and fieldwork by the authors (e.g., Fu et al. [19])

Genetic clustering of three Gentiana species based on genomic SNPs. (A) Results of principal coordinate analysis. (B) Bar plots showing probabilities of ancestral clusters of each sample with K = 3 in Admixture
Phylogenetic relationship and hybridization among species
Genomic SNPs data resulted in a well-supported tree (Fig. 4). In general, samples clustered together according to the species they were attributed to, except for one population (Fu2016070) of G. lawrencei, which was sister to G. veitchiorum. Gentiana hexaphylla diverged first, and thus was the sister lineages to both G. lawrencei and G. veitchiorum (Fig. 4).

(A) Heatmap of weighted Weir and Cockerham’s FST; (B) Genetic distance (FST/(1- FST)) against geolographical distance between populations of three Gentiana species of section Kudoa
Patterson’s D-statistic revealed strong signals of introgression between G. lawrencei and the other two Gentiana species (Fig. 5). The DBBAA values between G. lawrencei and G. hexaphylla showed that introgression was detected in most population pairs between the two species with p < 1 × 10− 6 (below a Bonferroni-adjusted P-value threshold of 0.001). The signal of introgression between most population pairs was also detected in populations of G. lawrencei and G. veitchiorum (Fig. 5). Week signal of introgression was observed between G. hexaphylla and G. veitchiorum (Fig. 5).

Maximum likelihood tree of three Gentiana species based on genomic SNPs. Phylogenetic support values for maximum likelihood were shown above branches only when they differ from 100% bootstrap support. Codes in the tips indicated the population names
Relationship between genomic differentiation and environmental variables
When all the 19 climate variables were analyzed, 16 out of 19 variables (except bio5, bio10 and bio14) showed significant differences between at least two of the three gentians (Fig. 6). The 16 variables could be grouped into two categories related to temperature (bio1–bio4, bio6-bio9, bio11) and precipitation (bio12–bio13, bio15–bio19). Gentiana lawrencei showed significant differences with the other two gentians in bio1, bio4, bio6–bio9, bio11, bio12, bio17 and bio19. One variable (bio12; Annual Precipitation) showed significant difference among the three gentians, namely G. hexaphylla, G. veitchiorum and G. lawrencei preferred higher, intermediate, and relatively lower annual mean precipitation, respectively (Fig. 6).

Gene flow detected in the three Gentiana species. Heatmap summarizing the D-statistic (DBBAA) estimates and their P-values. Taxa P2 and P3 are displayed on the x- and y-axes. Each square represents the highest estimate of each combination of P2 and P3 population. The colour of each square signifies the D-statistic estimate. D-statistic tests for which p < 1×10-6 (i.e. were below a Bonferroni-adjusted P-value threshold of 0.001) are marked with a black asterisk. The white squares in the matrix indicate no data
After the Pearson correlation analysis, 10 climatic variables (bio1-bio6, bio12, bio14, bio15, bio17) were kept for the analysis. The results of RDA analysis showed that the 10 combined bioclimatic variables had variables contribution in the first two principal components (Fig. 7). Seven variables, bio2 (Mean Diurnal Range), bio3 (Isothermality), bio4 (Temperature Seasonality), bio6 (Min Temperature of Coldest Month), bio14 (Precipitation of Driest Month), bio15 (Precipitation Seasonality) and bio17 (Precipitation of Driest Quarter) had a more substantial contribution on the first principal component (PC1, 25.06%), and the remaining variables on the second principal component (PC2, 15.05%). Among the variables, bio3, bio4, bio6, bio12 (Annual Precipitation), bio14, bio15 and bio17 were significantly differed (P < 0.05, Table 2). Gentiana hexaphylla differed with G. veitchiorum and G. lawrencei along PC1, and the latter two species differed along PC2. For G. hexaphylla and the another two gentians, the variables that explained this differentiation were related to temperature and precipitation in months outside of the growing season, for example bio6 (Min Temperature of Coldest Month), bio14 (Precipitation of Driest Month) and bio17 (Precipitation of Driest Quarter) (Fig. 7). For G. veitchiorum and G. lawrencei, the variables that explained the differentiation were mainly related to bio5 (Max Temperature of Warmest Month).

Comparison of climate variables among three Gentiana species. Climate variables had no significant difference were not shown. ***, P < 0.001; **, P < 0.01; *, P < 0.05; NS, no significant. Bio1, annual mean temperature; Bio2, Mean Diurnal Range; Bio3, isothermality; Bio4, Temperature Seasonality; Bio6, min temperature of coldest month; Bio7, Temperature Annual Range; Bio8, Mean Temperature of Wettest Quarter; Bio9, Mean Temperature of Driest Quarter; Bio11, Mean Temperature of Coldest Quarter; Bio12, annual precipitation; Bio13, Precipitation of Wettest Month; Bio15, Precipitation Seasonality; Bio16, Precipitation of Wettest Quarter; Bio17, Precipitation of Driest Quarter; Bio18, Precipitation of Warmest Quarter; Bio19, Precipitation of Coldest Quarter
Discussion
Genetic divergence among the threeGentianaspecies.
By sampling G. hexaphylla, G. veitchiorum and G. lawrencei across populations known to be spatially structured and thus building upon previous studies [19, 38, 44], our genomic data showed clear genetic divergence among the three sympatric gentians (Fig. 2). The three species are distinct genetic entities, as supported by several morphological traits such as opposite or whorl leaves, shape of leaves, corolla and calyx lobes, as well as corolla color [27, 36]. Nevertheless, the differentiation among the three species was not very strong as the FST value between species ranged from 0.149 to 0.243. We observed that the genetic divergence within species was sometimes larger than that between species and was sometimes associated with geographical distances (Fig. 3). Interestingly, a weak but significant positive correlation was detected between interspecific genetic distance and geological distance (Fig. 3B), rather than the expected negative correlation [45, 46]. This shows the impact of geographical scale on interspecific divergence in the three gentians. Our findings suggest that both physical barriers and heterogeneous environments may have caused isolation and strengthened differentiation among the three Gentiana species. Indeed, previous studies showed that intra-species genetic geographical patterns in G. veitchiorum and G. lawrencei were shaped by isolation in a southeastern and a northwestern refugia [19, 44], whereas the pattern in G. hexaphylla was mainly shaped by geological features at the center of the HM [38]. Genomic data showed that adaption to heterogeneous environments in the QTP region could have produced a high intraspecific divergence [FST=0.89, 20] and a parallel adaptive divergence in a number plants taxa [23]. Therefore, we suggest that geographical isolation as well as adaption to heterogeneous environments could have fostered the differentiation among the three Gentiana species, and thus had a profound impact in their divergence.
Ancient hybridization and polyploidization versus divergence
With a remarkable number of radiations occurring in the alpine biome of the region of the QTP [6], of which many closely related species are sympatric, it is now crucial to gather more evidence on the role of hybridization and introgression for adaptation, speciation and ultimately diversification. Our analyses showed obvious gene flow among the three gentians (Figs. 2 and 5), which should come as no surprise since hybridization appears to be more common than previously thought in Gentiana. Indeed, it was observed in at least two regions of the world, including the QTP region [e.g., 38–40] and Europe (listed in Favre et al. [25]), as well as in different sections of Gentiana (e.g., sect. Ciminalis, sect. Gentiana, sect. Cruciata, etc.). A more frequent occurrence of hybridization in Gentiana would also explain some major challenges encountered in taxonomical work and species identification within the genus, as for example in section Chondrophyllae. This is also the case for several taxa of G. section Kudoa, where continuous values for some morphological diagnostic traits were observed [27, 36]. In this section, hybridization may even occur among more species, in this case suggesting a reticulate evolution in its infancy.

Redundancy analysis showed the relationship between the independent climate variables and genetic divergence among three Gentiana species. The colored points represented individuals of the three species
In our study, signatures of gene flow were detected especially between G. lawrencei towards both G. hexaphylla and G. veitchiorum in almost all populations (Fig. 5), indicating that their history of hybridization could date back to the origin of these species. Supporting this fact is for example the higher genetic diversity index in G. lawrencei, and the lower FST value between G. lawrencei and the other two species. In fact, previous studies have already detected some cases of hybridization between G. lawrencei and G. veitchiorum in a refugium shared by the two species [19]. Furthermore, species distribution models showed that the three species have had a large overlap of potential distribution through time, which increased since the LIG [19, 38], rendering hybridization and introgression progressively more likely in the last few thousand years.
Hybridization may be followed by allopolyploidization, sometimes setting up the base for the emergence of new taxa, as seen for example in birch trees [47]. Whether this has been the case in section Kudoa or elsewhere in the genus is still difficult to verify, but it is possible since 30.43% of Gentiana species (28 out of 92) for which karyological data are available are in fact polyploids. For the hybridizing species investigated here, karyological studies showed that G. veitchiorum is diploid (2n = 2x = 24, [32, 34]), whereas G. lawrencei is tetraploid (2n = 4x = 48, [33, 34]). In this study, we estimated that the respective genome sizes of G. veitchiorum, G. lawrencei and G. hexaphylla are 3.25 G, 5.00 G and 3.18 G. These results thus suggest that G. hexaphylla – of which ploidy level is still technically unknown - is likely to be a diploid, possibly with ca. 24 chromosomes. Including G. hexaphylla, two out of six species in section Kudoa are tetraploids [32,33,34,35]. Taking their background of hybridization into account, it is not impossible that allopolyploidization participated in the divergence of the three Kudoa species investigated in this study. Yet, a more complete investigation on hybridization and allopolyploidization including all species of that section may be needed.
Did climatic preferences contribute to divergence?
When comparing climatic preferences of G. hexaphylla, G. veitchiorum and G. lawrencei, we found significant differences regarding some components of precipitation and temperature. For example, G. hexaphylla and G. veitchiorum appear to occur in areas with more precipitation, higher temperature, and smaller temperature annual range than G. lawrencei (Fig. 6). In particular, G. hexaphylla prefers habitats with more precipitation than the other two species, consistent with its range limited to the HM, which are characterized by a wetter climate than other areas in the QTP region. Our phylogenetic analysis showed that G. veitchiorum and G. lawrencei diverged from G. hexaphylla (Fig. 4), and that divergence among G. hexaphylla, G. veitchiorum and G. lawrencei occurred in the Pleistocene [31], in parallel with the local climate becoming progressively cooler and drier [56], each sample was digested with the restriction enzyme EcoRI, followed by ligation, purification and size selection as described in Fu et al. [38]. Paired-end reads 150 bp in length were generated using the Illumina Novaseq 6000 (Tian**, China). Raw reads were filtered and trimmed with Trimmomatic v0.32 [57] with default parameters to remove adaptor sequences and low-quality reads and sites, and then checked for quality with FastQC v0.11.2.
Since RAD-seq study employing reference-based approaches was recommended [58], we mapped the raw reads against the chromosome-level genome of Gentiana dahurica (PRJNA799480; [59]), the closest available genome, with bwa-men v2.2.1 [60], and produced the sequence alignment/map format files with SAMtools v1.9 [61]. We marked the PCR duplications with sambamba v0.8.1 [62], and called variations with freebayes v0.9.21 [63] with default parameters. Only SNPs were retained in vcftools 0.1.13 [64] for downstream analysis. SNPs with a minor allele frequency (MAF, less than 5%) and missing frequency of more than 0.8 among individuals were removed using vcftools v0.1.13 [64]. Linkage-disequilibrium SNP pruning was performed in vcftools to excludes variants from each pair closer than 100 bp. We used PGDSpider v2.1.1.5 [65] to convert the final VCF file into different formats to perform further analyses.
Genetic diversity and population genetic structure
We computed allelic richness (Ar), observed heterozygosity (HO), gene diveristy (HS), and Wright’s inbreeding coefficient (FIS) using the R package hierfstat [66]. To assess the levels of genetic differentiation among populations, we estimated pairwise FST among populations using the Weir and Cockerham method [67] in R package hierfstat [66]. A Mantel test was performed in R v. 4.0.1 [68] to check the correlation between genetic diversity (Ar and HO) and altitude in each species. The pairwise FST was graphically displayed with package pheatmap (https://cran.r-project.org/web/packages/pheatmap/) using R. We plotted FST/(1-FST) against pairwise geographic distances among populations to illustrate the range-wide isolation by distance (IBD) pattern in R.
For exploring the genetic clusters, we used a Bayesian clustering method implemented in Admixture [69] based on the identified SNPs, with assumed clusters (K) from 1 to 20. The cross-validation (CV) procedure performed 10-fold CV (--cv = 10). The CV errors were plotted in R to assess the model complexity for the data. Graphical representation of individual cluster assignments was performed using DISTRUCT v1.1 [70]. The same data set was used to perform a principal component analysis (PCA), with ten main principal components (PCs) extracted in PLINK v1.90 [71], and visualized using R.
Phylogenetic analysis
We constructed a phylogenetic tree based on the genomic SNPs using maximum likelihood (ML) in IQ-TREE v.1.6.8 [72] with 1000 ultrafast bootstraps [73]. The best-fitted substitution model was chosen in ModelFinder v2 [74]. The Python script ‘vcf2phylip’ (https://github.com/edgardomortiz/vcf2phylip) was used to transfer the SNPs data for tree building. Gentiana waltonii (specimen no. Fu2020030) was served as the outgroup.
Hybridization analysis
We tested for introgression among the three Gentiana species using Patterson’s D-statistic [75]. The D-statistic uses asymmetry in gene tree topologies to quantify introgression between either of two lineages which share a common ancestor (P1 and P2) and one other lineage (P3) that diverged from the common ancestor of P1 and P2 earlier. We calculated Patterson’s D-statistic for all possible population trios using the Dtrios function of Dsuite v0.5.r44 [76] with default parameters. We fixed G. waltonii as the outgroup. We assessed significance of each test using 100 jackknife resampling runs, and visualized the D-statistic estimates in R.
Environmental data analysis
We obtained 19 climate variables based on the average values from 1950 to 2000 for G. hexaphylla, G. veitchiorum and G. lawrencei from WorldClim (https://www.worldclim.org) using the R package raster [77] at 30 arc-second resolutions. Significant differences among the three species for these environmental factors were visualized in R.
Highly correlated variables (Pearson’s correlation coefficient > 0.8, p < 0.01) were detected in SPSS and removed to reduce the number of predictors. We used the function rda from the R package vegan [78] to perform the redundancy analysis (RDA) in order to identify potential environmental factors driving genomic differentiation. We used the function anova.cca from the R package vegan to check the significance of each predictor.
Data availability
The dataset supporting the conclusions of this article is available in the Figshare (doi: https://doi.org/10.6084/m9.figshare.20445879). The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [79] in National Genomics Data Center [80], China National Center for Bioinformation/Bei**g Institute of Genomics, Chinese Academy of Sciences (GSA: CRA008165) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
References
Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403(6772):853–8. https://doi.org/10.1038/35002501.
Antonelli A, Kissling WD, Flantua SG, Bermúdez MA, Mulch A, Muellner-Riehl AN, et al. Geological and climatic influences on mountain biodiversity. Nat Geosci. 2018;11(10):718–25. https://doi.org/10.1038/s41561-018-0236-z.
Mosbrugger V, Favre A, Muellner-Riehl AN, Päckert M, Mulch A. Cenozoic evolution of geo-biodiversity in the Tibeto-Himalayan region. In: Hoorn C, Perrigo A, Antonelli A. Mountains, Climate, and Biodiversity. Hoboken: John Wiley & Sons; 2018. pp. 429–48.
Noroozi J, Talebi A, Doostmohammadi M, Rumpf SB, Linder HP, Schneeweiss GM. Hotspots within a global biodiversity hotspot–areas of endemism are associated with high mountain ranges. Sci Rep. 2018;8:10345. https://doi.org/10.1038/s41598-018-28504-9.
Badgley C, Smiley TM, Terry R, Davis EB, DeSantis LR, Fox DL, et al. Biodiversity and topographic complexity: modern and geohistorical perspectives. Trends Ecol Evol. 2017;32(3):211–26. https://doi.org/10.1016/j.tree.2016.12.010.
Muellner-Riehl AN, Schnitzler J, Kissling WD, Mosbrugger V, Rijsdijk KF, Seijmonsbergen AC, et al. Origins of global mountain plant biodiversity: Testing the ‘mountain‐geobiodiversity hypothesis’. J Biogeogr. 2019;46(12):2826–38. https://doi.org/10.1111/jbi.13715.
Darwin C. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London: John Murray; 1859.
Savolainen O, Lascoux M, Merilä J. Ecological genomics of local adaptation. Nat Rev Genet. 2013;14(11):807–20. https://doi.org/10.1038/nrg3522.
Hawkins BA, Field R, Cornell HV, Currie DJ, Guégan JF, Kaufman DM,.et al. Energy, water, and broad-scale geographic patterns of species richness. Ecology. 2003;84(12):3105–17. https://doi.org/10.1890/03-8006.
Kreft H, Jetz W. Global patterns and determinants of vascular plant diversity. P Natl A Sci. 2007;104(14):5925–30. https://doi.org/10.1073/pnas.0608361104.
Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405(6789):907–13. https://doi.org/10.1038/35016000.
Spieth PT. Gene flow and genetic differentiation. Genetics. 1974;78:961.
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N, et al. Hybridization and speciation. J Evol Biol. 2013;26(2):229–46. https://doi.org/10.1111/j.1420-9101.2012.02599.x.
Sun H, Zhang J, Deng T, Boufford DE. Origins and evolution of plant diversity in the Hengduan Mountains, China. Plant Divers. 2017;39(4):161. https://doi.org/10.1016/j.pld.2017.09.004.
Ding WN, Ree RH, Spicer RA, **ng YW. Ancient orogenic and monsoon-driven assembly of the world’s richest temperate alpine flora. Science. 2020;369(6503):578–81. https://doi.org/10.1126/science.abb4484.
Tang H, Micheels A, Eronen JT, Ahrens B, Fortelius M. Asynchronous responses of East Asian and Indian summer monsoons to mountain uplift shown by regional climate modelling experiments. Clim Dynam. 2013;40:1531–49. https://doi.org/10.1007/s00382-012-1603-x.
Favre A, Packert M, Pauls SU, Jahnig SC, Uhl D, Michalak I, et al. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol Rev. 2015;90:236–53. https://doi.org/10.1111/brv.12107.
Li Q, Sun H, BouffordDE, Bartholomew B, FritschPW, Chen J, et al. Grade of Membership models reveal geographical and environmental correlates of floristic structure in a temperate biodiversity hotspot. New Phytol. 2021;232:1424–35. https://doi.org/10.1111/nph.17443.
Fu PC, Sun SS, Khan G, Dong XX, Tan JZ, Favre A, et al. Population subdivision and hybridization in a species complex of Gentiana in the Qinghai-Tibetan Plateau. Ann Bot. 2020;125:677–90. https://doi.org/10.1093/aob/mcaa003.
Zhang X, Sun Y, Landis JB, Zhang J, Yang L, Lin N, et al. Genomic insights into adaptation to heterogeneous environments for the ancient relictual Circaeaster agrestis (Circaeasteraceae, Ranunculales). New Phytol. 2020;228(1):285–301. https://doi.org/10.1111/nph.16669.
Liu JQ, Duan YW, Hao G, Ge XJ, Sun H. Evolutionary history and underlying adaptation of alpine plants on the Qinghai-Tibet Plateau. J Syst Evol. 2014;52:241–9. https://doi.org/10.1111/jse.12094.
Zhao W, Sun YQ, Pan J, Sullivan AR, Arnold ML, Mao JF, Wang XR. Effects of landscapes and range expansion on population structure and local adaptation. New Phytol. 2020;228(1):330–43. https://doi.org/10.1111/nph.16619.
Wang Z, Jiang Y, Yang X, Bi H, Li J, Mao X, et al. Molecular signatures of parallel adaptive divergence causing reproductive isolation and speciation across two genera. Innovation. 2022;3(3):100247. https://doi.org/10.1016/j.xinn.2022.100247.
Wu S, Wang Y, Wang Z, Shrestha N, Liu J. Species divergence with gene flow and hybrid speciation on the Qinghai-Tibet Plateau. New Phytol. 2022;234(2):392–404. https://doi.org/10.1111/nph.17956.
Favre A, Paule J, Ebersbach J. Incongruences between nuclear and plastid phylogenies challenge the identification of correlates of diversification in Gentiana in the European alpine system. Alp Bot. 2021;132:29–50. https://doi.org/10.1007/s00035-021-00267-6.
Ebersbach J, Tkach N, Röser M, Favre A. The role of hybridisation in the making of the species-rich arctic-alpine genus Saxifraga (Saxifragaceae). Diversity, 2020:12(11):440. https://doi.org/10.3390/d12110440.
Ho TN, Liu SW. A worldwide monograph of Gentiana. Bei**g: Science Press; 2001.
Favre A, Michalak I, Chen CH, Wang JC, Pringle JS, Matuszak S, et al. Out-of-Tibet: the spatio-temporal evolution of Gentiana. (Gentianaceae) J Biogeogr. 2016;43:1967–78. https://doi.org/10.1111/jbi.12840.
Favre A, Pringle JS, Heckenhauer J, Kozuharova E, Gao Q, Lemmon EM, et al. Phylogenetic relationships and sectional delineation within Gentiana (Gentianaceae). Taxon. 2020;69(6):1221–1238. https://doi.org/10.1002/tax.12405.
Sun SS, Fu PC, Zhou XJ, Cheng YW, Zhang FQ, Chen SL, et al. The complete plastome sequences of seven species in Gentiana sect. Kudoa (Gentianaceae): insights into plastid gene loss and molecular evolution. Front Plant Sci. 2018;9:493. https://doi.org/10.3389/fpls.2018.00493.
Fu PC, Sun SS, Twyford AD, Li BB, Zhou RQ, Chen SL, et al. Lineage-specific plastid degradation in subtribe Gentianinae (Gentianaceae). Ecol. Evol. 2021;11:3286–99. https://doi.org/10.1002/ece3.7281.
Yuan YM. Karyological studies on Gentiana section Cruciata Gaudin (Gentianaceae) from China. Caryologia. 1993;46(2–3):99–114.
Yuan YM, Küpfer P. Karyological studies of Gentianopsis Ma and some related genera of Gentianaceae from China. Cytologia. 1993;58(2):115–23.
Chen SL, He TN, Liu JQ. The chromosome number of eight species in Gentiana (Gentianaceae) from alpine mountains of the western China. Acta Bot Boreal -Occident Sin. 1997;17(4):547–50.
Ho TN, Liu JQ, Chen SL. Contribution to the karyomorphology of 7 species in Gentiana (Gentianaceae). Acta Biol Plateau Sin. 2002;15:67–75.
Ho TN, Pringle JS. Gentiana. In: Wu ZY, Raven PH, eds. Flora of China, vol. 16. Bei**g: Science Press; St. Louis: Missouri Botanical Garden. 1995;15–97.
Hou QZ, Duan YW, Si QW, Yang HL. Pollination ecology of Gentiana lawrencei var. farreri, a late-flowering Qinghai-Tibet Plateau species. Chin J Plant Ecol. 2019;33(6):1156. https://doi.org/10.3773/j.issn.1005-264x.2009.06.016.
Fu PC, Sun SS, Hollingsworth PM, Chen SL, Favre A, Twyford AD. Population genomics reveal deep divergence and strong geographical structuring in the Hengduan Mountains. Front Plant Sci. 2022. https://doi.org/10.3389/fpls.2022.936761.
Hu Q, Peng H, Bi H, Lu Z, Wan D, Wang Q, Mao KS. Genetic homogenization of the nuclear ITS loci across two morphologically distinct gentians in their overlap** distributions in the Qinghai-Tibet Plateau. Scie Rep. 2016;6:34244. https://doi.org/10.1038/srep34244.
Fu PC, Twyford AD, Sun SS, Wang HY, **a MZ, Tan CX, et al. Recurrent hybridization underlies the evolution of novelty in Gentiana (Gentianaceae) in the Qinghai-Tibetan Plateau. AoB Plants. 2021;13:plaa068. https://doi.org/10.1093/aobpla/plaa068.
Sun SS, Guo YL, Favre A, Fu PC. Genetic differentiation and evolutionary history of two medicinal gentians (Gentiana stipitata Edgew. and Gentiana szechenyii Kanitz) in the Qinghai-Tibet Plateau. J Appl Res Med Arom. 2022;30:100375. https://doi.org/10.1016/j.jarmap.2022.100375.
Haemmerli M. Molecular aspects in systematics of “Gentiana” sect. “Calathianae” Froel. Université de Neuchâtel. 2007.
Christe C, Caetano S, Aeschimann D, Kropf M, Diadema K, Naciri Y. The intraspecific genetic variability of siliceous and calcareous Gentiana species is shaped by contrasting demographic and re-colonization processes. Mol Phylogenet Evol. 2014;70:323–36. https://doi.org/10.1016/j.ympev.2013.09.022.
Fu PC, Ya HY, Liu QW, Cai HM, Chen SL. Out of refugia: population genetic structure and evolutionary history of the alpine medicinal plant Gentiana lawrencei var. farreri (Gentianaceae). Front. Genet. 2018;9:564. https://doi.org/10.3389/fgene.2018.00564.
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, et al. The effect of geographical scale of sampling on DNA barcoding. Syst Biol. 2012;61(5):851–69. https://doi.org/10.1093/sysbio/sys037.
Chen W, Hubert N, Li Y, **ang D, Cai X, Zhu S, et al. Large scale DNA barcoding of the subfamily Culterinae (Cypriniformes: Xenocyprididae) in East Asia unveils geographic scale effect, taxonomic warnings and cryptic diversity. Mol Ecol. 2022;31(14):3871–87. https://doi.org/10.1111/mec.16540.
Wang N, Kelly LJ, McAllister HA, Zohren J, Buggs RJ. Resolving phylogeny and polyploid parentage using genus-wide genome-wide sequence data from birch trees. Mol Phylogenet Evol. 2021;160:107126. https://doi.org/10.1016/j.ympev.2021.107126.
Farnsworth A, Lunt DJ, Robinson SA, Valdes PJ, Roberts WH, Clift PD, et al. Past East Asian monsoon evolution controlled by paleogeography, not CO2. Sci. Adv. 2019;5(10):eaax1697. https://doi.org/10.1126/sciadv.aax16.
Deng T, Wu F, Wang S, Su T, Zhou Z. Major turnover of biotas across the Oligocene/Miocene boundary on the Tibetan Plateau. Palaeogeogr Palaeocl. 2021;567:110241. https://doi.org/10.1016/j.palaeo.2021.110241.
Ahrens CW, ByrneM, Rymer PD. Standing genomic variation within coding and regulatory regions contributes to the adaptive capacity to climate in a foundation tree species. Mol Ecol. 2019;28(10):2502–16. https://doi.org/10.1111/mec.15092.
Jordan R, Hoffmann AA, Dillon SK, Prober SM. Evidence of genomic adaptation to climate in Eucalyptus microcarpa: Implications for adaptive potential to projected climate change. Mol Ecol. 2017;26(21):6002–20. https://doi.org/10.1111/mec.14341.
Pepin N, Bradley R, Diaz H, Baraer M, Caceres EB, Forsythe N, et al. Elevation-dependent warming in mountain regions of the world. Nat Clim Change. 2015;5:424–30. https://doi.org/10.1038/nclimate2563.
Yang L, Li H, Li Q, Guo Q, Li J. Genetic diversity analysis and potential distribution prediction of Sophora moorcroftiana endemic to Qinghai–Tibet Plateau, China. Forests. 2021;12(8):1106.
Guo Q, Li H, Zheng W, Pan J, Lu J, Li J, Zheng Y. Analysis of genetic diversity and prediction of Larix species distribution in the Qinghai–Tibet Plateau, China. J Forestry Res. 2022; 1–11. https://doi.org/10.1007/s11676-022-01513-1.
Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc. 2007;2(9):2233–44. https://doi.org/10.1038/nprot.2007.310.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genoty** using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–8. https://doi.org/10.1101/gr.5681207.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Shafer AB, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, et al. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol Evol. 2017;8(8):907–17. https://doi.org/10.1111/2041-210X.12700.
Li T, Yu X, Ren Y, Kang M, Yang W, Feng L, et al. The chromosome-level genome assembly of Gentiana dahurica (Gentianaceae) provides insights into gentiopicroside biosynthesis. DNA Res. 2022;29(2):dsac008. https://doi.org/10.1093/dnares/dsac008.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4. https://doi.org/10.1093/bioinformatics/btv098.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. ar**v preprint ar**v. 2012.1207.3907. https://doi.org/10.48550/ar**v.1207.3907.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. https://doi.org/10.1093/bioinformatics/btr330.
Lischer HE, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28(2):298–9. https://doi.org/10.1093/bioinformatics/btr642.
Goudet J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes. 2005;5:184–6. https://doi.org/10.1111/j.1471-8286.2004.00828.x.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70. https://doi.org/10.2307/2408641.
Core Team R R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. https://www.R-project.org/.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. https://doi.org/10.1101/gr.094052.109.
Rosenberg NE. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8. https://doi.org/10.1046/j.1471-8286.2003.00566.x.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Amer J Human Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. https://doi.org/10.1093/molbev/msu300.
Hoang DT, Chernomor O, Von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35(2):518–22. https://doi.org/10.1093/molbev/msx281.
Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587. https://doi.org/10.1038/nmeth.4285.
Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–52. https://doi.org/10.1093/molbev/msr048.
Malinsky M, Matschiner M, Svardal H. Dsuite-fast D‐statistics and related admixture evidence from VCF files. Mol Ecol Resour. 2021;21(2):584–95. https://doi.org/10.1111/1755-0998.13265.
Hijmans RJ, Van Etten J, Cheng J, Mattiuzzi M, Sumner M, Greenberg JA, et al. Package ‘raster’. R package, 2015;734.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara RB, et al. Package ‘vegan’. Community Ecology Package, version, 2.0. 2013.
The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom Proteom Bioinf. 2021;19(4):578–83. https://doi.org/10.1016/j.gpb.2021.08.001.
Database Resources of the National Genomics Data Center. China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022;50(D1):D27–38. https://doi.org/10.1093/nar/gkab951.
Acknowledgements
We are grateful to Zhizhong Li of Wuhan Botanical Garden, Chinese Academy of Sciences for help in data analysis, and to Shulin Zhang of Henan University to offer cotton seeds in genome size estimation.
Funding
This work was financially supported by the Foundation of Educational Commission of Henan Province [22A180024] to S.S, the National Natural Science Foundation of China [31600296] to P.F.
Author information
Authors and Affiliations
Contributions
P.F. designed the experiments, collected the samples, analysed the data and wrote the main manuscript; A.F. revised the manuscript; R.W and Y.H. participated in the bioinformatic analysis; S.S. designed the experiments, analysed the data and revised the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We comply with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora and confirm that all methods were performed in accordance with the relevant guidelines/regulations/legislation. These wild species samples in this study have not been included in the list of key protected plants. The sampling of wild species in this study was met local policy requirements and did not affect the survival or reproduction of the species. The planting material were identified by Dr. Pengcheng Fu. Voucher specimens were deposited in the herbarium of Luoyang Normal University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, P., Favre, A., Wang, R. et al. Between allopatry and secondary contact: differentiation and hybridization among three sympatric Gentiana species in the Qinghai-Tibet Plateau. BMC Plant Biol 22, 504 (2022). https://doi.org/10.1186/s12870-022-03879-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03879-0