
Abstract
Background
Atriplex canescens is a typical C4 secretohalophyte with salt bladders on the leaves. Accumulating excessive Na+ in tissues and salt bladders, maintaining intracellular K+ homeostasis and increasing leaf organic solutes are crucial for A. canescens survival in harsh saline environments, and enhanced photosynthetic activity and water balance promote its adaptation to salt. However, the molecular basis for these physiological mechanisms is poorly understood. Four-week-old A. canescens seedlings were treated with 100 mM NaCl for 6 and 24 h, and differentially expressed genes in leaves and roots were identified, respectively, with Illumina sequencing.
Results
In A. canescens treated with 100 mM NaCl, the transcripts of genes encoding transporters/channels for important nutrient elements, which affect growth under salinity, significantly increased, and genes involved in exclusion, uptake and vacuolar compartmentalization of Na+ in leaves might play vital roles in Na+ accumulation in salt bladders. Moreover, NaCl treatment upregulated the transcripts of key genes related to leaf organic osmolytes synthesis, which are conducive to osmotic adjustment. Correspondingly, aquaporin-encoding genes in leaves showed increased transcripts under NaCl treatment, which might facilitate water balance maintenance of A. canescens seedlings in a low water potential condition. Additionally, the transcripts of many genes involved in photosynthetic electron transport and the C4 pathway was rapidly induced, while other genes related to chlorophyll biosynthesis, electron transport and C3 carbon fixation were later upregulated by 100 mM NaCl.
Conclusions
We identified many important candidate genes involved in the primary physiological mechanisms of A. canescens salt tolerance. This study provides excellent gene resources for genetic improvement of salt tolerance of important crops and forages.
Similar content being viewed by others

Background
Salinity is one of the most severe abiotic factors threatening agricultural productivity and ecological environment throughout the world [2, 12]. Approximately half of irrigated lands in the world are threatened by salinity [43]. The expansion of soil salinization and the increasing human population are forcing agricultural production into marginal areas [44]. Soil salinization can significantly reduce the yield and quality of crops by resulting in a series of metabolic disturbances due to ionic toxicity, physiological drought and nutrient deficiency [14, 63]. Therefore, improving the salt tolerance of plants, especially traditional crops and forages, is currently an urgent issue, since most of these species are glycophytes with weak salt tolerance [43, 51]. In contrast, halophytic species have evolved multiple adaptation strategies to deal with harsh saline environments [12, 44]. Learning from halophytes, understanding the mechanisms underlying plant response to salinity and thereby identifying key genes related to salt tolerance will contribute to breeding crops with salt tolerance [61].
Halophytes can be divided into three types based on their adaptive strategies to saline environments: (i) pseudohalophytes maintain a low Na+ level by limiting Na+ uptake [52], (ii) euhalophytes compartmentalize Na+ into swollen internal vacuoles to alleviate Na+ toxicity in the cytosol [14, 56] and (iii) secretohalophytes exclude excessive Na+ from secreting structures (salt glands or salt bladders) on the surface of stems and/or leaves [11, 44]. For example, Reaumuria trigyna and Limonium bicolor are capable of secreting Na+ via their multicellular salt glands, but excreting little K+ to maintain high K+/Na+ ratio in the shoots [8, 62]. Mesembryanthemum crystallinum, Chenopodium quinoa and Atriplex species deposit a large amount of Na+ in epidermal bladder cells (EBCs) to improve salt tolerance of plants [26, 39]. Approximately half of all halophyte plants possess salt bladders, which segregate excessive Na+ away from metabolically active organs in the growing plant body [13, 44]; hence, these plants are likely to be potential species for saline soil amelioration and improvement of salt tolerance in important crops.
Atriplex canescens (four-wing saltbush), a C4 perennial semi-evergreen woody shrub with excellent adaptability to salinity and drought, is a typical secretohalophyte with salt bladders that is widely distributed in saline and arid regions [22]. This species is commonly planted in highway medians and on road shoulders, slopes, and other disturbed areas for erosion control and reclamation of marginal lands, and it can be used as a landscape plant in the arid regions of northern China; moreover, A. canescens is an attractive fodder crop for most livestock because of its high palatability and nutritional value [17, 40]. Early research findings showed that A. canescens could grow along a salinity gradient from 72 to 2017 mol/m3 NaCl in the root zone and accumulated more Na+ than K+ for osmotic adjustment (OA) at relatively low salinities [17, 18]. Our previous study revealed that moderate salinity (100 mM NaCl) could stimulate the growth of A. canescens and high salinity (400 mM NaCl) had no significant effect on its growth [40]. Under saline conditions, A. canescens can enhance photosynthetic capacity, accumulate more Na+ in tissues and salt bladders, maintain leaf K+ homeostasis, and use inorganic ions as well as organic osmolytes for OA, which may contribute to water balance in the plant [40]. Our latest investigation showed that the addition of 100 mM NaCl effectively alleviated the adverse impact of drought on the growth of A. canescens by increasing the accumulation of solutes (Na+, free proline, betaine and soluble sugar) in leaves as well as the net photosynthetic rate and water content (Guo H. and Bao A.K., unpublished data). All of these results indicate that the transport of Na+ and K+, the accumulation of organic solutes, the improvement of photosynthetic activity and leaf hydration are vital strategies for A. canescens adaptation to saline environments. Nonetheless, the possible molecular basis of these important physiological mechanisms is poorly understood owing to the absence of genomic data in A. canescens.
High-throughput RNA sequencing has been widely used to investigate the molecular processes related to adaptive responses to abiotic stresses and to identify stress-resistance candidate genes by analyzing differences in transcript abundance [57]. In this work, transcriptomes of A. canescens were generated by Illumina assembly technology to lay the foundation for exploring the potential salt tolerance mechanisms of this species. In addition, the genes showing significant transcriptional changes in A. canescens under NaCl treatment were then identified by comparing the gene transcript profiles in leaves and roots between salt-treated and control plants by using a tag-based digital gene expression (DGE) system, mainly focusing on identifying the candidate genes related to ion transport, organic osmolyte accumulation, water transport and photosynthesis.
Results
Transcriptome sequencing, de novo assembly and unigene functional annotation
A total of 13.37 and 13.41 Gb clean bases were generated from the leaves and roots of A. canescens by Illumina HiSeq sequencing, respectively (Additional file 1: Table S1). Then, 207.20 Mb raw reads were yielded from leaves and 210.00 Mb raw reads were yielded from roots through high-throughput sequencing (Additional file 1: Table S1). After filtering, a total of 133.70 and 134.12 Mb clean reads were generated from the leaves and roots, respectively, coupled with a Q20 score greater than 97 and 0.00% Ns (Additional file 1: Table S1). All of these results indicated that the output and quality of transcriptome sequencing were adequate for subsequent analysis.
Paired-end information was used to join contigs into scaffolds and further assembly23, and 54,611 and 59,582 unigenes, with a mean length of 912 and 696 bp, respectively, were generated from the leaves and roots (Table 1). Then, 70,571 all-unigene sequences were acquired, with a mean length of 961 bp, N50 of 1647 bp and GC percentage of 40.01%, after further assembly of the unigenes from leaves and roots (Table 1). The size distribution is shown in Additional file 1: Figure S1, and the lengths of 24,205 unigenes were more than 1000 bp.
Then, 44,121 unigenes (62.52% of the 70,571 unigenes) were annotated to known genes in 7 databases, namely, the Nr, Nt, Swiss-Prot, KEGG, COG, InterPro and GO databases (Additional file 1: Table S2). Functional annotation was not obtained for 37.48% of the unigenes due to the absence of genomic data in A. canescens and close-related species. Among these annotated unigenes, 26,021 unigenes annotated in the COG database in terms of sequence homology were classified into 25 functional clusters and 37,395 unigenes annotated with GO terms were grouped into 3 main GO categories with 52 subcategories (Additional file 1:Figures S2 and S3).
Differentially expressed genes (DEGs) in A. canescens under NaCl treatment
Eight independent cDNA libraries (CL6, CR6, SL6, SR6, CL24, CR24, SL24 and SR24) were sequenced, and approximately 22 million raw reads were generated in each library; after filtering low-quality reads, we obtained 21 million clean reads in each library (data not shown), more than 72% of which could be mapped to the transcriptome reference database (data not shown).
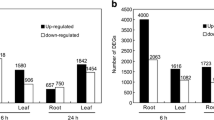
The DEGs in A. canescens were analyzed by comparing the 100 mM NaCl treatment with the control. When plants were subjected to the 100 mM NaCl treatment for 6 h, 14,686 and 16,306 DEGs were found in the leaves and roots, respectively, using the thresholds of FDR < 0.001 and |log2Ratio| > 1 (Fig. 1). Among these DEGs, 9023 and 4824 DEGs were upregulated, including 1768 and 1031 DEGs that were expressed in the leaves and roots of plants in the 100 mM NaCl treatment but almost not expressed in the control (the FPKM value was 0.01 in control plants, the same below), respectively (Fig. 1). Interestingly, the number of upregulated DEGs was much greater than that of downregulated DEGs in the leaves; conversely, the number of upregulated DEGs was much lower than that of downregulated DEGs in the roots. In addition, 3403 and 2405 DEGs (including 1799 and 1196 upregulated DEGs) were identified in the leaves and roots of plants treated to 100 mM NaCl for 24 h, respectively, which was far fewer than the number identified in plants exposed to the treatment for 6 h. Among these upregulated DEGs, 629 and 426 DEGs were expressed in the leaves and roots of plants in the 100 mM NaCl treatment while almost not expressed in the control, respectively (Fig. 1).

Numbers of differentially expressed genes (DEGs) in leaves and roots of A. canescens under 100 mM NaCl for 6 and 24 h. A FDR < 0.001 and an absolute value of the log2Ratio > 1 were used as the thresholds to determine significant differences in gene expression
DEGs related to ion, glucose and oxygen transport
Firstly, the DEGs related to ion transport especially Na+ transport into salt bladders were identified under 100 mM NaCl treatment since accumulating a great quantity of Na+ in salt bladders and maintaining intracellular ion homeostasis are vital strategies for A. canescens adaptation to saline conditions.
In leaves, the number of upregulated DEGs associated with ion transport was much larger than that of downregulated DEGs when plants were treated with 100 mM NaCl for either 6 h or 24 h (Fig. 2). After plants were treated with 100 mM NaCl for 6 h, 76 DEGs were upregulated, which included important transcripts related to Na+ (including NHX and HKT), K+ (such as AKT and SKOR), Ca2+ (CNGC, CCX and P-Ca2+ ATPase), Mg2+ (MGT), and NH4+ (AMT) transport and the anion transport of NO3− (NRT), PO43− (PHT), SO42− (STAS), Cl− (CLC and SLAH) and several important micronutrients (such as Zn, Mo, B and Cu) (Fig. 2a; Additional file 1: Table S3). Some genes encoding plasma membrane H+-ATPases (P-H+ ATPase) and vacuolar H+-pyrophosphatases (V-H+ PPase) were also upregulated (Fig. 2a). The number of DEGs was significantly lower under 100 mM NaCl for 24 h than under 100 mM NaCl for 6 h (Fig. 2b). Among these upregulated genes, two transcripts associated with Na+ transport (SOS1 and HKT1) were upregulated, and the other upregulated DEGs were mainly SKOR, AKT, CNGC, MGT and NRT transport protein family genes, which are related to nutrient element transport (Fig. 2b; Additional file 1: Table S4). In addition, among these DEGs related to K+ transport in leaves, 9 DEGs were significantly upregulated under 100 mM NaCl but not under control conditions for either 6 or 24 h (Table 2).

The DEGs related to ion transport in leaves of A. canescens under 100 mM NaCl for 6 (a) and 24 h (b). NHX: tonoplast Na+/H+ antiporter, SOS1: plasma membrane Na+/H+ antiporter, HKT: high-affinity K+ transporter, KEA: K+ efflux antiporter, KCO: calcium-activated outwardly rectifying potassium channel, SKOR: stelar K+ outwardly rectifying channel, AKT: inwardly rectifying K+ channel, KT/HAK/KUP: K+ transporter, CNGC: cyclic nucleotide-gated channel, CCX: cation/Ca2+ exchanger, P-Ca2+ ATPase: plasma membrane Ca2+ ATPase, V-CAX: vacuolar cation/H+ exchanger, P-H+ ATPase: plasma membrane H+ ATPase, V-H+ PPase: vacuolar H+ PPase, MGT: Mg2+ transporter, AMT: NH4+ transporter, NRT: NO3− transporter, STAS: SO42− transporter, PHT: PO43− transporter, CLC: vacuolar Cl−/H+ exchanger, SLAH: Slow type anion channel, CTR: Cu2+ transporter, BOR: BO3− transporter, ZnT: Zn3+ transporter, MOT: MoO42− transporter. The up and down arrows indicate the total number of up and downregulated DEGs, respectively. The same below
The epidermal bladder cell (EBC) together with stalk cell (SC) and epidermal cell (EC) constitute the EC-SC-EBC complex [44]. The Na+ sequestration in the high vacuolization of salt bladder is achieved by four times Na+ transport through plasma membrane and one time Na+ transport through tonoplast [37]; furthermore, tomato regulates the activity of these enzymes by controlling the expression patterns of their genes to adapt to salinity [30]. Amylase (AMS), mannitol dehydrogenase (MD) and trehalose-6-phosphate synthase (TPS) play indispensable roles in starch, mannitol and trehalose metabolism, respectively, and their enzyme activities are positively related to the salt tolerance of plants [7]. In this study, multiple genes encoding key enzymes for the synthesis of proline, betaine and soluble sugar in leaves were upregulated under 100 mM NaCl (Fig. 5; Additional file 1: Tables S7-S8), and 13 genes (such as P5CS, BADH and SPS) of these upregulated genes were continuously expressed under salt treatment for both 6 and 24 h (Table 3), suggesting that A. canescens possesses an efficient mechanism for accumulating osmoprotectants under saline conditions by modulating the expression patterns of important genes involved in compatible solute biosynthesis, which would be conducive to protecting plants in saline environments by OA.
Aquaporin plays an important role in the regulation of water balance in A. canescens under saline conditions
A. canescens seedlings can maintain a higher leaf relative water content by effective OA under salinity treatment [40]. Moreover, our latest study found that water was abundantly transported as a solvent into salt bladders with the accumulation of Na+ in salt bladders of A. canescens under NaCl treatment, causing rapid expansion of the salt bladders, a sharp increase in turgor pressure and eventually the bursting of the bladders and subsequent release of a large amount of accumulated Na+ (Guo H. and Bao A.K., unpublished data); this finding suggests that the rapid accumulation of water in salt bladders is the key factor affecting salt secretion in A. canescens. AQPs can effectively regulate the water balance inside and outside of the plant cell by specifically mediating the rapid transmembrane transport of water [6]. In this study, many DEGs related to AQPs were upregulated in leaves but not in roots under 100 mM NaCl for 6 h, including 6 nodulin-like intrinsic proteins (NIPs), 1 small basic intrinsic protein (SIP), 2 plasma membrane intrinsic proteins (PIPs) and 3 tonoplast intrinsic proteins (TIPs) (Table 4). The NIPs mainly mediate boron uptake (NIP5;1) or are involved in pollen development and pollination (NIP4) in Arabidopsis thaliana [10, 55]. The SIPs are localized to the endoplasmic reticulum membrane but currently are not well characterized [25]. Plasma membrane-located PIPs are divided into the PIP1 and PIP2 groups; PIP2 members mainly function as water channels, while PIP1 members usually have much lower or no water conductivity due to their failure to localize to the plasma membrane [5]. Our results showed that the transcript of 1 PIP gene (CL969.Contig2_All, highly homologous to PIP2;3) was significantly upregulated in leaves (Table 4). PIP2 in Malus zumi Mats is involved in water movement during both water absorption and transport and alters the salt tolerance of transgenic Arabidopsis [53]. An AQP protein, AcPIP2, characterized from A. canescens, improved plant growth rate and salt tolerance when overexpressed in A. thaliana [28]. Moreover, tonoplast-located TIPs, primarily mediating the accumulation of water in the vacuole, play vital roles in maintaining cell turgor and enhancing the capacity for OA and are also able to indirectly promote Na+ compartmentation into vacuoles, which is conducive to improving plant adaptation to saline environments [25, 36]. In the present study, we found that the transcripts of 3 TIPs (Unigene15728_All, highly homologous to TIP2;2; Unigene12348_All, highly homologous to TIP4;1 and CL5261.Contig2_All, highly homologous to TIP1;3) were sharply upregulated in the leaves but downregulated in the roots of A. canescens under NaCl treatment, and there was even a 14.24-fold increase in the transcript level of AcTIP2;2 (Table 4). Therefore, the AcPIP2 and AcTIPs in the leaves of A. canescens are likely to be involved in the transport of water into salt bladders under salt treatment, which facilitates salt secretion and the maintenance of the water balance in leaves and might result from the accumulation of solutes in leaf tissues and salt bladders.
Moderate salinity improves photosynthesis of A. canescens by increasing the transcripts of photosynthesis-related genes
Previous studies showed that NaCl significantly improved the photosynthetic capacity of A. canescens plants, and the trends of photosynthetic indicators were different from those of C3 xerohalophytes, suggesting that Na+ may promote the C4 photosynthetic process of A. canescens under saline conditions [40]. C4 plants share stronger CO2 assimilation capacity and can sufficiently utilize light energy; moreover, this kind of plant have greater adaptability to adversity since they can take advantage of low CO2 levels in the intercellular space under stress conditions [15, 27]. The oxygenic photosynthesis of higher plants can be divided into three stages: the primary reaction, photosynthetic electron transport and photophosphorylation and CO2 assimilation. The first two steps in this process involve the conversion of sunlight into active chemical energy, which is driven by several multisubunit membrane protein complexes, including photosystem II, cytochrome b6/f, photosystem I, ferredoxin and ATP synthase [38]. The last reaction is a process that converts CO2 into stable chemical energy stored in organic matter by using the energy (ATP and NADPH) produced by the light reaction [15]. Our study found that the majority of DEGs related to the above-mentioned complexes and the enzymes involved in carbon fixation and chlorophyll biosynthesis/catabolism were downregulated under 100 mM NaCl for 6 h (Fig. 7a, Additional file 1: S13), but the few upregulated genes were mainly involved in electron transport and carbon fixation, the latter of which were almost always involved in the C4 pathway, including phosphoenolpyruvate carboxylase (PEPC), malate dehydrogenase (MDH), malic enzyme (ME) and aspartate aminotransferase (AST) (Table 5). These enzymes play key roles in the C4 photosynthetic pathway and are capable of providing more CO2 for the C3 pathway in the vascular bundle sheath61. Interestingly, the transcript levels of many DEGs involved in the processes of chlorophyll biosynthesis, electron transport and carbon fixation (based on the C3 pathway) were significantly upregulated after treatment with 100 mM NaCl for 24 h but not after treatment for 6 h (Table 5), suggesting that A. canescens preferentially increased the transcript abundances of genes encoding key enzymes in the C4 pathway to improve its assimilation capacity and then increased the transcript levels of other genes encoding complexes related to chlorophyll biosynthesis, electron transport and C3 carbon fixation under salt treatment, which might be one of the important reasons the photosynthesis of A. canescens was significantly improved by 100 mM NaCl [40]. At present, re-engineering C3 plants with C4 CO2-concentrating mechanisms is of broad interest [34, 45]. Overexpression of plastidic ZmNADP-MDH (NADP-MDH) in maize conferred salt tolerance to transgenic Arabidopsis [24]. Thus, the results of this study provide abundant genetic resources for improving photosynthetic efficiency in C3 crops/forages through genetic engineering.
Conclusions
This study identified candidate genes showing significant transcriptional changes in A. canescens under 100 mM NaCl treatment, mainly focusing on genes related to ion transport, organic osmolyte synthesis, water transport and photosynthesis. The abundance of transcripts encoding transporters/channels for important macro- and microelements was significantly increased by 100 mM NaCl, which is conducive to promote the uptake and transport of nutrient elements. It is worth noting that some genes related to Na+ transport in leaves (such as AcSOS1, AcHKT1 and AcNHX) might play crucial roles in the excretion of salt via epidermis bladder cells. In addition, the transcripts of a number of genes related to the synthesis of organic osmolytes in leaves was significantly upregulated by NaCl treatment, which allowed the accumulation of more organic solutes to enhance OA under salt treatment. Moreover, 100 mM NaCl promoted water transport in A. canescens by inducing the transcripts of aquaporin-encoding genes in leaves. Interestingly, NaCl preferentially induced the transcripts of genes encoding proteins participating in the C4 photosynthetic pathway to provide greater assimilation capacity for photosynthesis and then increased the transcript levels of other genes encoding complexes related to chlorophyll biosynthesis, electron transport and C3 carbon fixation under salt treatment. Our results lay the foundation for investigating molecular mechanisms of salt tolerance in secretohalophytes and provide a theoretical basis for genetic improvement of stress tolerance in important crops and forages by using the outstanding gene resources from A. canescens.
Methods
Plant materials and experimental treatments
Seeds of Atriplex canescens were collected from Lingwu County in Ningxia Autonomous Region, China. After removed the hard seed coat with 75% H2SO4 (v/v) for 15 h, the seeds were washed many times with purified water until the washings has no smell and then germinated in moist vermiculite at 28 °C in the dark for 5 days. Uniform seedlings were transplanted into plastic containers (5 cm × 5 cm × 5 cm; two plants/pot) filled with vermiculite and irrigated with 1/2-strength Hoagland nutrient solution at 2-day intervals [40]. Plants were cultured at 28 °C/25 °C (day/night), 16/8 h photoperiod (light/dark; the light density was approximately 800 μmol/m2/s) and 65% relative humidity.
Four-week-old seedlings were treated with 1/2-strength Hoagland nutrient solution supplemented with 0 (control) or 100 mM NaCl. The leaves and roots of seedlings in the two treatments were collected after treatment for 6 and 24 h, respectively. A total of eight samples were marked as follows: CL6, CR6, SL6, SR6, CL24, CR24, SL24 and SR24; C and S represent the control and treatment with 100 mM NaCl, respectively; 6 and 24 denote the treatment duration; and R and L denote the roots and leaves, respectively. For example, SL6 and SR6 were the leaf and root samples, respectively, from salt-treated plants for which seedlings were treated for 6 h. All the fresh samples were immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
RNA preparation, cDNA library construction and Illumina sequencing
Total RNA was isolated from the eight samples with an RNeasy Plant Mini Kit (Qiagen). The extracted RNA was quantified by using a NanoDrop ND-1000 instrument (Thermo Scientific), and the integrity of the RNA was determined by 1% agarose gel electrophoresis. Equivalent amounts of total RNA isolated from each of the four leaf tissues (CL6, SL6, CL24 and SL24) and each of the four corresponding root tissues (CR6, SR6, CR24 and SR24) were pooled. The two mRNA pools were then used for reverse transcription to obtain two cDNA libraries as the cDNA in the leaves and roots of A. canescens by using the method described by Dang et al. [8] and sequenced on an Illumina HiSeq™ 2000 platform in BGI Shenzhen.
De novo assembly and functional annotation
High-quality clean reads were created after filtering adaptor sequences, duplicated sequences, reads containing more than 5% ambiguous bases (‘N’) and reads in which more than 50% of bases showed a Q-value ≤5. After filtering out low quality reads, de novo assembly was proceeded by using Trinity; then, the Trinity unigenes were clustered with TGICL software to minimize sequence redundancy [21]. The unigenes were divided into two classes after performing gene family clustering, one class included clusters with the prefix CL contained several unigenes with a sequence similarity of more than 70%, and the other included singletons with the prefix unigenes [31]. To attach predicted gene informations for assembled unigenes, the sequences were functionally annotated based on seven protein databases (including the Nr, Nt, Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), InterPro and Gene Ontology (GO) databases) using the BLASTX tool with an E-value ≤10− 5 threshold. Blast2GO software was employed to perform functional categorization by GO terms on the basis of biological process, cellular component and molecular function ontologies, and the Web Gene Ontology Annotation Plot (WEGO) tool was used to statistically analyze the data [8].
Differentially expressed gene (DEG) library preparation and analysis
Eight independent cDNA libraries (CL6, CR6, SL6, SR6, CL24, CR24, SL24 and SR24) were prepared in parallel for leaves and roots at different times under salt treatment by using a tag-based DGE kit [59]. Then, each library was sequenced through the Illumina HiSeq™ 2000 sequencing platform in BGI Shenzhen. After low-quality reads (including reads with adaptors, more than 10% unknown nucleotides (‘N’) and only one copy number) were removed, the clean reads were mapped to the transcriptome reference database. And then, the transcript levels of all assembled unigenes were calculated by using the number of fragments per kb per million reads (FPKM) method to identify differentially expressed genes (DEGs) [59]. In addition, the false discovery rate (FDR) method was used to confirm the threshold P-value for multiple tests and analysis by manipulating the FDR value. An FDR < 0.001 and an absolute value of |log2Ratio| > 1 were used as thresholds to identify DEGs [8].
qRT-PCR validation of DEGs
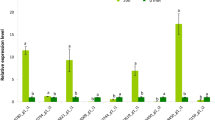
To experimentally evaluate the RNA-Seq results, total RNA was extracted from the 8 samples as described before and reversely transcribed into cDNA according to the manufacturer’s protocol (TaKaRa Biotechnology). The qRT-PCR was conducted by using SYBR Green Real-Time PCR Master Mix (TaKaRa Biotechnology) and performed on a StepOnePlus Real-Time PCR Thermocycler (Applied Biosystems, USA). ACTIN gene was used as the internal standard. The relative transcript levels of the 30 randomly selected unigenes were calculated using the 2-ΔΔCt method [31].
Abbreviations
- AKT:
-
Inwardly rectifying K+ channel
- AMS:
-
Amylase
- AMT:
-
NH4+ transporter
- BADH:
-
Betaine aldehyde dehydrogenase
- BOR:
-
BO3− transporter
- CCX:
-
Cation/Ca2+ exchanger
- CLC:
-
Vacuolar Cl−/H+ exchanger
- CMO:
-
Choline monooxygenase
- CNGC:
-
Cyclic nucleotide-gated channel
- COG:
-
Clusters of orthologous groups
- CTR:
-
Cu2+ transporter
- DEG:
-
Differentially expressed gene
- EBC:
-
Epidermal bladder cell
- EC:
-
Epidermal cell
- FDR:
-
False discovery rate
- FPKM:
-
Number of transcripts per million clean reads
- GDH:
-
Glutamate dehydrogenase
- GLUT:
-
Glucose transporter
- GO:
-
Gene ontology
- GOGAT:
-
Gluamate synthase
- HB:
-
Hemoglobin
- HKT:
-
High-affinity K+ transporter
- INV:
-
Invertase
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- KT/HAK/KUP:
-
K+ transporter
- MD:
-
Mannitol dehydrogenase
- MGT:
-
Mg2+ transporter
- MOT:
-
MoO42− transporter
- NHX:
-
Tonoplast Na+/H+ antiporter
- NIP:
-
Nnodulin-like intrinsic protein
- NRT:
-
NO3− transpoter
- OAT:
-
Ornithine aminotransferase
- P5CS:
-
Δ1-pyrroline-5-carboxylate synthetase
- P-Ca2+ ATPase:
-
Plasma membrane Ca2+ ATPase
- PEAMT:
-
Phosphoethanolamine N-methyltransferase
- P-H+ ATPase:
-
Plasma membrane H+ ATPase
- PHT:
-
PO43− transporter
- PIP:
-
Plasma membrane intrinsic protein
- SC:
-
Stalk cell
- SIP:
-
Small basic intrinsic protein
- SKOR:
-
Stelar K+ outward rectifying channel
- SLAH:
-
Slow type anion channel
- SOS1:
-
Plasma membrane Na+/H+ antiporter
- SPS:
-
Sucrose-phosphate synthase
- SS:
-
Starch synthase
- STAS:
-
SO42− transporter
- SuSy:
-
Sucrose synthase
- TIP:
-
Tonoplast intrinsic protein
- TPS:
-
Trehalose-phosphate synthase
- V-CAX:
-
Vacuolar cation/H+ exchanger
- V-H+ PPase:
-
Vacuolar H+ PPase
- ZnT:
-
Zn3+ transporter
References
Aranda-Sicilia MN, Cagnac O, Chanroj S, Sze H, Rodriguez-Rosales MP, Venema K. Arabidopsis KEA2, a homolog of bacterial KefC, encodes a K+/H+ antiporter with a chloroplast transit peptide. Biochim Biophys Acta. 2012;1818:2362–71.
Bao AK, Du BQ, Touil L, Kang P, Wang QL, Wang SM. Co-expression of tonoplast cation/H+ antiporter and H+-pyrophosphatase from xerophyte Zygophyllum xanthoxylum improves alfalfa plant growth under salinity, drought and field conditions. Plant Biotechnol J. 2016;14:964–75.
Barratt DHP, Derbyshire P, Findlay K, Pike M, Wellner N, Lunn J, Feil R, Simpson C, Maule AJ, Smith AM. Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc Natl Acad Sci U S A. 2009;106:13124–9.
Böhm J, Messerer M, Müller HM, Scholz-Starke J, Gradogna A, Scherzer S, Maierhofer T, Bazihizina N, Zhang H, Stigloher C, Ache P, Al-Rasheid KAS, Mayer KFX, Shabala S, Carpaneto A, Haberer G, Zhu JK, Hedrich R. Understanding the molecular basis of salt sequestration in epidermal bladder cells of Chenopodium quinoa. Curr Biol. 2018;28:1–11.
Chaumont F, Barrieu F, Jung R, Chrispeels MJ. Plasma membrane intrinsic proteins from maize cluster in two sequence subgroups with differential aquaporin activity. Plant Physiol. 2000;122:1025–34.
Chaumont F, Tyerman SD. Aquaporins: highly regulated channels controlling plant water relations. Plant Physiol. 2014;164:1600–18.
Conde A, Regalado A, Rodrigues D, Costa JM, Blumwald E, Chaves MM, Gerós H. Polyols in grape berry: transport and metabolic adjustments as a physiological strategy for water-deficit stress tolerance in grapevine. J Exp Bot. 2015;66:889–906.
Dang ZH, Zheng LL, Wang J, Gao Z, Wu SB, Qi Z, Wang YC. Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genomics. 2013;14:29.
Davenport RJ, Munoz-Mayor A, Jha D, Essah PA, Rus A, Tester M. The Na+ transporter AtHKT1 controls retrieval of Na+ from the xylem in Arabidopsis. Plant Cell Environ. 2007;30:497–507.
Di Giorgio JAP, Bienert GP, Ayub ND, Yaneff A, Barberini ML, Mecchia MA, Amodeo G, Soto GC, Muschiettia JP. Pollen-specific aquaporins NIP4;1 and NIP4;2 are required for pollen development and pollination in Arabidopsis thaliana. Plant Cell. 2016;28:1053–77.
Ding F, Yang JC, Yuan F, Wang BS. Progress in mechanism of salt excretion in recretohalopytes. Front Biol. 2010;5:164–70.
Flowers TJ, Colmer TD. Plant salt tolerance: adaptations in halophytes. Ann Bot. 2015;115:327–31.
Flowers TJ, Galal HK, Bromham L. Evolution of halophytes: multiple origins of salt tolerance in land plants. Funct Plant Biol. 2010;37:604–12.
Flowers TJ, Munns R, Colmer TD. Sodium chloride toxicity and the cellular basis of salt tolerance in halophytes. Ann Bot. 2015;115:419–31.
Furbank RT. Evolution of the C4 photosynthetic mechanism: are there really three C4 acid decarboxylation types? J Exp Bot. 2011;62:3103–8.
Gaymard F, Pilot G, Lacombe B, Bouchez D, Bruneau D, Boucherez J, Michaux-Ferrière N, Thibaud J, Sentenac H. Identification and disruption of a plant shaker-like outward channel involved in K+ release into the xylem sap. Cell. 1998;94:647–55.
Glenn EP, Olsen M, Frye R, Moore D, Miyamoto S. How much sodium accumulation is necessary for salt tolerance in subspecies of the halophyte Atriplex canescens? Plant Cell Environ. 1994;17:711–9.
Glenn EP, Pfister R, Brown JJ, Thompson TL, O'Leary J. Na and K accumulation and salt tolerance of Atriplex canescens (Chenopodiaceae) genotypes. Am J Bot. 1996;83:997–1005.
Gobert A, Isayenkov S, Voelker C, Czempinski K, Maathuis FJ. The two-pore channel TPK1 gene encodes the vacuolar K+ conductance and plays a role in K+ homeostasis. Proc Natl Acad Sci U S A. 2007;104:10726–31.
Gobert A, Park G, Amtmann A, Sanders D, Maathuis FJM. Arabidopsis thaliana cyclic nucleotide gated channel 3 forms a nonselective ion transporter involved in germination and cation transport. J Exp Bot. 2006;57:791–800.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen ZH, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma FD, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Hao GY, Lucero ME, Sanderson SC, Zacharias EH, Holbrook NM. Polyploidy enhances the occupation of heterogeneous environments through hydraulic related trade-offs in Atriplex canescens (Chenopodiaceae). New Phytol. 2013;197:970–8.
** YK, **g W, Zhang Q, Zhang WH. Cyclic nucleotide gated channel 10 negatively regulates salt tolerance by mediating Na+ transport in Arabidopsis. J Plant Res. 2015;128:211–20.
Kandoi D, Mohanty S, Tripathy BC. Overexpression of plastidic maize NADP-malate dehydrogenase (ZmNADP-MDH) in Arabidopsis thaliana confers tolerance to salt stress. Protoplasma. 2017;255:547–63.
Kayum MA, Park JI, Nath UK, Biswas MK, Kim HT, Nou IS. Genome-wide expression profiling of aquaporin genes confer responses to abiotic and biotic stresses in Brassica rapa. BMC Plant Biol. 2017;17:23.
Kiani-Pouya A, Roessner U, Jayasinghe NS, Lutz A, Rupasinghe T, Bazihizina N, Bohm J, Alharbi S, Hedrich R, Shabala S. Epidermal bladder cells confer salinity stress tolerance in the halophyte quinoa and Atriplex species. Plant Cell Environ. 2017;40:1900–15.
Kustka AB, Milligan AJ, Zheng H, New AM, Gates C, Bidle KD, Reinfelder JR. Low CO2 results in a rearrangement of carbon metabolism to support C4 photosynthetic carbon assimilation in Thalassiosira pseudonana. New Phytol. 2015;204:507–20.
Li JT, Yu G, Sun XH, Liu YZ, Liu JL, Zhang XH, Jia CG, Pan HY. AcPIP2, a plasma membrane intrinsic protein from halophyte Atriplex canescens, enhances plant growth rate and abiotic stress tolerance when overexpressed in Arabidopsis thaliana. Plant Cell Rep. 2015;34:1401–15.
Liu K, Li LG, Luan S. Intracellular K+ sensing of SKOR, a shaker-type K+ channel from Arabidopsis. Plant J. 2006;46:260–8.
Lu SW, Li TL, Jiang J. Effects of salinity on sucrose metabolism during tomato fruit development. Afr J Biotechnol. 2010;9:842–9.
Ma Q, Bao AK, Chai WW, Wang WY, Zhang JL, Li YX, Wang SM. Transcriptomic analysis of the succulent xerophyte Zygophyllum xanthoxylum, in response to salt treatment and osmotic stress. Plant Soil. 2016a;402:343–61.
Ma Q, Hu J, Zhou XR, Yuan HJ, Kumar T, Luan S, Wang SM. ZxAKT1 is essential for K+ uptake and K+/Na+ homeostasis in the succulent xerophyte Zygophyllum xanthoxylum. Plant J. 2016b;90:48–60.
Ma Q, Li YX, Yuan HJ, Hu J, Wei L, Bao AK, Zang JL, Wang SM. ZxSOS1 is essential for long-distance transport and spatial distribution of Na+ and K+ in the xerophyte Zygophyllum xanthoxylum. Plant Soil. 2014;374:661–76.
Mackinder LCM. The chlamydomonas CO2-concentrating mechanism and its potential for engineering photosynthesis in plants. New Phytol. 2018;217:54–61.
Masclaux-Daubresse, Reisdorf-Cren M, Pageau K, Lelandais M, Grandjean O, Kronenberger J, Valadier MH, Feraud M, Jouglet T, Suzuki A. Glutamine synthetase-glutamate synthase pathway and glutamate dehydrogenase play distinct roles in the sink-source nitrogen cycle in tobacco. Plant Physiol. 2006;140:444–56.
Maurel C, Verdoucq L, Rodrigues O. Aquaporins and plant transpiration. Plant Cell Environ. 2016;39:2580–7.
Miron D, Schaffer AA. Sucrose phosphate synthase, sucrose synthase, and invertase activities in develo** fruit of Lycopersicon esculentum mill. And the sucrose accumulating Lycopersicon hirsutum Humb. And Bonpl. Plant Physiol. 1991;95:623–7.
Nevo R, Charuvi D, Tsabari O, Reich Z. Composition, architecture and dynamics of the photosynthetic apparatus in higher plants. Plant J. 2012;70:157–76.
Oh DH, Barkla BJ, Vera-Estrella R, Pantoja O, Lee SY, Bohnert HJ, Dassanayake M. Cell type-specific responses to salinity − the epidermal bladder cell transcriptome of Mesembryanthemum crystallinum. New Phytol. 2015;207:627–44.
Pan YQ, Guo H, Wang SM, Zhao BY, Zhang JL, Ma Q, Yin HJ, Bao AK. The photosynthesis, Na+/K+ homeostasis and osmotic adjustment of Atriplex canescens in response to salinity. Front Plant Sci. 2016;7:848.
Qin D, Zhao CL, Liu XY, Wang PW. Transgenic soybeans expressing betaine aldehyde dehydrogenase from Atriplex canescens show increased drought tolerance. Plant Breed. 2017;136:699–709.
Sahoo DP, Kumar S, Mishra S, Kobayashi Y, Panda SK, Sahoo L. Enhanced salinity tolerance in transgenic mungbean overexpressing Arabidopsis antiporter (NHX1) gene. Mol Breeding. 2016;36:144.
Shabala S. Learning from halophytes: physiological basis and strategies to improve abiotic stress tolerance in crops. Ann Bot. 2013;112:1209–21.
Shabala S, Bose J, Hedrich R. Salt bladders: do they matter? Trends Plant Sci. 2014;19:687–91.
Sharwood RE, Ghannoum O, Whitney SM. Prospects for improving CO2 fixation in C3-crops through understanding C4-rubisco biogenesis and catalytic diversity. Curr Opin Plant Biol. 2016;31:135–42.
Shi HZ, Ishitani M, Kim C, Zhu JK. The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc Natl Acad Sci U S A. 2000;97:6896–901.
Shi HZ, Quintero FJ, Pardo JM, Zhu JK. The putative plasma membrane Na+/H+ antiporter SOS1 controls long-distance Na+ transport in plants. Plant Cell. 2002;14:465–77.
Shirasawa K, Takabe T, Takabe T, Kishitani S. Accumulation of glycinebetaine in rice plants that overexpress choline monooxygenase from spinach and evaluation of their tolerance to abiotic stress. Ann Bot. 2006;98:565–71.
Sunarpi HT, Motoda J, Kubo M, Yang H, Yoda K, Horie R, Chan WY, Leung HY, Hattori K, Konomi M, Osumi M, Yamagami M, Schroeder JI, Uozumi N. Enhanced salt tolerance mediated by AtHKT1 transporter-induced Na+ unloading from xylem vessels to xylem parenchyma cells. Plant J. 2005;44:928–38.
Székely G, Abrahám E, Cséplo A, Rigó G, Zsigmond L, Csiszár J, Ayaydin F, Strizhov N, Jásik J, Schmelzer E, Koncz C, Szabados L. Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 2008;53:11–28.
Tang XL, Mu XM, Shao HB, Wang HY, Brestic M. Global plant-responding mechanisms to salt stress: physiological and molecular levels and implications in biotechnology. Crit Rev Biotechnol. 2015;35:425–37.
Wang B, Davenport RJ, Volkov V, Amtmann A. Low unidirectional sodium influx into root cells restricts net sodium accumulation in Thellungiella halophila, a salt-tolerant relative of Arabidopsis thaliana. J Exp Bot. 2006;57:1161–70.
Wang L, Li QT, Lei Q, Feng C, Gao YN, Zheng XD, Zhao Y, Wang Z, Kong J. MzPIP2;1: an aquaporin involved in radial water movement in both water uptake and transportation, altered the drought and salt tolerance of transgenic Arabidopsis. PLoS One. 2015;10:e0142446.
Wang P, Li ZW, Wei JS, Zhao ZL, Sun DY, Cui SJ. A Na+/Ca2+ exchanger-like protein (AtNCL) involved in salt stress in Arabidopsis. J Biol Chem. 2012;287:44062–70.
Wang SL, Yoshinari A, Shimada T, Hara-Nishimura I, Mitani-Ueno N, Ma JF, Naito S, Takano J. Polar localization of the NIP5;1 boric acid channel is maintained by endocytosis and facilitates boron transport in Arabidopsis roots. Plant Cell. 2017;29:824–42.
Wang SM, Zhang JL, Flowers TJ. Low affinity Na+ uptake in the halophyte Suaeda maritima. Plant Physiol. 2007;145:559–71.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63.
Wei C, Cui Q, Zhang XQ, Zhao YQ, Jia GX. Three P5CS genes including a novel one from Lilium regale play distinct roles in osmotic, drought and salt stress tolerance. J Plant Biol. 2016;59:456–66.
Xue J, Bao YY, Li BL, Cheng YB, Peng ZY, Liu H, Xu HJ, Zhu ZR, Lou YG, Cheng JA, Zhang CX. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PLoS One. 2010;5:e14233.
Yang TY, Zhang S, Hu YB, Wu FC, Hu QD, Chen G, Cai J, Wu T, Moran N, Yu L, Xu GH. The role of a potassium transporter OsHAK5 in potassium acquisition and transport from roots to shoots in rice at low potassium supply levels. Plant Physiol. 2014;166:945–59.
Yang Z, Zheng HX, Wei XC, Song J, Wang BS, Sui N. Transcriptome analysis of sweet sorghum inbred lines differing in salt tolerance provides novel insights into salt exclusion by roots. Plant Soil. 2018;430:423–39.
Yuan F, Lyu MJA, Leng BY, Zhu XG, Wang BS. The transcriptome of NaCl-treated Limonium bicolor leaves reveals the genes controlling salt secretion of salt gland. Plant Mol Biol. 2016;91:241–56.
Zhang JL, Shi HZ. Physiological and molecular mechanisms of plant salt tolerance. Photosynth Res. 2013;115:1–22.
Zhu M, Shabala L, Cuin TA, Huang X, Zhou M, Munns R, Shabala S. Nax loci affect SOS1-like Na+/H+ exchanger expression and activity in wheat. J Exp Bot. 2016;67:835–44.
Zou CS, Chen AJ, **ao LH, Muller HM, Ache P, Haberer G, Zhang ML, Jia W, Deng P, Huang R, Lang D, Li F, Zhan DL, Wu XY, Zhang H, Bohm J, Liu RY, Shabala S, Hedrich R, Zhu JK, Zhang H. A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res. 2017;27:1327–40.
Acknowledgements
We thank all the members in our laboratory for providing useful discussions. We are very grateful to Dr. Shelley Roanne Hepworth from Carleton University, Canada, for valuable suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (31730093; 31670405), the National Key Research and Development Program of China (2017YFC0504804), and the Fundamental Research Funds for the Central Universities (lzujbky-2018-k01). Funds were used for the design of the study, collection, analysis, and interpretation of data and in writing the manuscript, as well as in the open access payment.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
A-KB and HG conceived and designed the study. HG and LZ performed the experiments. HG, LZ and Y-NC collected and analyzed data. HG wrote the manuscript. S-MW, A-KB and Y-NC revised the manuscript. All author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1
: Figure S1. Length distribution of all assembled unigenes. Figure S2. COG function distribution of all unigenes. A total of 26,012 putative proteins showing significant homology to those in the COG database were classified into 25 functional clusters. X-axis indicates the number of unigenes in a cluster. Figure S3. GO function distribution of all unigenes. A total of 37,395 unigenes were assigned to GO terms and were summarized in 3 main GO categories and 52 subcategories. X-axis indicates the number of genes in a category. Figure S4. Correlation analysis for expression pattern validation of 30 randomly selected DEGs between RNA-Seq and qRT-PCR results. Table S1. Summary of sequencing reads after filtering. Table S2. Summary of sequence annotation. Table S3. Differentially expressed genes (DEGs) related to ion transport in leaves of A. canescens under 100 mM NaCl for 6 h. Table S4. DEGs related to ion transport in leaves of A. canescens under 100 mM NaCl for 24 h. Table S5. DEGs related to ion transport in roots of A. canescens under 100 mM NaCl for 6 h. Table S6. DEGs related to ion transport in roots of A. canescens under 100 mM NaCl for 24 h. Table S7. DEGs related to organic osmolytes synthesis in leaves of A. canescens under 100 mM NaCl for 6 h. Table S8. DEGs related to organic osmolytes synthesis in leaves of A. canescens under 100 mM NaCl for 24 h. Table S9. DEGs related to photosynthesis in leaves of A. canescens under 100 mM NaCl for 6 h. Table S10. DEGs related to photosynthesis in leaves of A. canescens under 100 mM NaCl for 24 h. Table S11. Expression pattern validation of 30 randomly selected genes in leaves of A. canescens under 100 mM NaCl for 6 and 24 h by qRT-PCR. Table S12. Expression pattern validation of 30 randomly selected genes in roots of A. canescens under 100 mM NaCl for 6 and 24 h by qRT-PCR. (DOCX 1081 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, H., Zhang, L., Cui, YN. et al. Identification of candidate genes related to salt tolerance of the secretohalophyte Atriplex canescens by transcriptomic analysis. BMC Plant Biol 19, 213 (2019). https://doi.org/10.1186/s12870-019-1827-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1827-6