
Abstract
Background
Flavonoids constitute a diverse class of secondary metabolites which exhibit potent bioactivities for human health and have been indicated to play an important role in plant development and defense. However, accumulation and variation of flavonoid content in diverse maize lines and the genes responsible for their biosynthesis in this important crop remain largely unknown. In this study, we combine genetic map**, metabolite profiling and gene regulatory network analysis to further enhance understanding of the maize flavonoid pathway.
Results
We repeatedly detected 25 QTL corresponding to 23 distinct flavonoids across different environments or populations. In addition, a total of 39 genes were revealed both by an expression based network analysis and genetic map**. Finally, the function of three candidate genes, including two UDP-glycosyltransferases (UGT) and an oxygenase which belongs to the flavone synthase super family, was revealed via preliminary molecular functional characterization.
Conclusion
We explored the genetic influences on the flavonoid biosynthesis based on integrating the genomic, transcriptomic and metabolomic information which provided a rich source of potential candidate genes. The integrated genomics based genetic map** strategy is highly efficient for defining the complexity of functional genetic variants and their respective regulatory networks as well as in hel** to select candidate genes and allelic variance before embarking on laborious transgenic validations.
Similar content being viewed by others


Background
Maize (Zea mays L.) is the world’s most widely grown crop for food, animal feed, biofuel and other industrial materials, and displays the highest global grain production [1]. By 2050, it is estimated that the human population will reach 9 billion [2]. Increasing yield while providing added nutritional value in maize is thus imperative to meet the growing nutritional demand of the huge global population [3, 40]. With the aid of high-throughput genoty** and metabolomics data, metabolic QTL were identified in a rice Zhenshan 97 and Minghui 63 recombinant inbred line (RIL) population, and some of the candidate genes for flavonoid content were further validated by examining over-expression transgenic rice lines [41]. A novel gene (BETA GLUCOSIDASE 6, BGLU6) was recently identified to be responsible for the production of flavonol 3-O-gentiobioside 7-O-rhamnoside (F3GG7R) in an Arabidopsis RIL population [42, 43]. In maize, hundreds of loci associated with metabolites from multiple pathways including flavonoid metabolism were identified, following genome wide association studies (GWAS) on a diverse maize population that revealing the genetic influences underlying metabolic variation [44]. In addition, near isogenic lines (NILs) containing P1-rr and P1-ww were used to study the co-expression and direct target genes of the R2R3-MYB transcription factor P1 [31]. Since P1 was proven to regulate some well-known genes involved in flavonoid biosynthesis, such as FLS1 and A1 through targeted molecular experiments [45], this study represented a great advance to systematically comprehend its gene regulatory circuitry. Maysin (C-glycosyl flavone) present in maize silks confers natural resistance to the corn earworm (Helicoverpa zea), which can cause severe damages on maize in the Americas [12]. Two loci that are capable of conferring salmon silks phenotypes, salmon silks 1 (sm1) and salmon silks 2 (sm2) were identified through QTL map** [30] in 2004. And previous genetic analyses predicted P1 to be epistatic to the salmon silk mutation [13]. Based on the available sm1 and sm2 map** information and knowledge of the genes regulated by P1 [13, 31], the molecular identification of the sm1 and sm2 gene products are revealed as a UDP-rhamnose synthase and a rhamnosyl transferase, respectively [12]. The molecular characterization of sm1 and sm2 therefore completes the maysin biosynthetic pathway. It can thus be anticipated that deep probing of further profiling studies will facilitate the elucidation of the genetic complexity of maize flavonoid biosynthesis. Indeed, integrative approaches are increasingly applied to enhance our understanding of metabolic pathway structure and regulation and how these affect the end-phenotypes of plants [46].
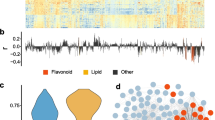
Previously, comprehensive metabolic profiling using liquid chromatography tandem mass spectrometry (LC-MS/MS) was carried out in mature maize kernels coming from several populations. Combined linkage analysis and GWAS was carried out on the resultant datasets which led to the identification of a variety of loci involved in multiple biosynthetic pathways [44, An association map** panel (AMP) and two RIL populations were planted in multiple environments (simply called AMPE1, AMPE2 for AMP, BBE1, BBE2 for BB RIL population, and ZYE1 and ZYE2 for ZY RIL population, which were described in detail in “Materials and Methods”) and the mature kernels harvested from these six field experiments were used for LC-MS/MS based metabolite profiling. In our previous metabolome-based GWAS study, 983 metabolite features were identified in the AMP [44]. 184 of these 983 metabolite features with chemical or putative annotations were analyzed in BB and ZY RIL populations subsequently [47]. In this study, we extract the profile of flavonoids from these previous datasets, which includes 29 flavonoids and five of them were chemically annotated. Briefly, these 29 flavonoids can be classified into flavones, flavanones, anthocyanins and methoxylated flavonoid. Among them, 28, 27, 23, 22, 25, 24 flavonoids were found in AMPE1, AMPE2, BBE1, BBE2, ZYE1, ZYE2, respectively, 15 flavonoids were detected in all the six environments (Table 1). The AMP and both RIL populations manifested great diversity in their flavonoid levels (Additional files 1 and 2: Tables S1 and S2), as indicated by the distribution of the log2 value of fold changes (Fig. 1a). In AMP, all flavonoids have broad-sense heritability (H2) greater than 0.3 and over 65% of flavonoids have H2 greater than 0.7. Over 45% and 60% of flavonoids have H2 greater than 0.5 in BB and ZY populations, respectively (Additional file 3: Figure S1). Correlation coefficient networks were also constructed based on flavonoid levels detected in each experiment, respectively, which demonstrated a clear separation between methoxylated flavonoids and other flavonoids, and most flavones were consistently linked to each other with R > 0.3 (Fig. 1b). Distribution of log2-fold changes and correlation coefficient based network of all flavonoids measured in AMP and two RIL populations. a Box plots showing the log2 value of fold changes of 29 flavonoids among the AMP and both BB and ZY RILs. Data from different environments (experiments) for AMP and each RIL population are shown. b Correlation coefficient based network of all flavonoids in each experiment for AMP and both BB and ZY populations. R ≥ 0.3 for correlation coefficient between two flavonoids was used to construct the network A total of 79 loci were identified by GWAS at significance level of P ≤ 1.8 × 10−6 in two experiments (AMPE1, AMPE2) (Table 2). Briefly, 51 loci were identified for 23 flavonoids in AMPE1, with an R2 (explained phenotypic variation) ranging from 6.84 to 19.77% and a mean of 8.93%; while 28 loci were detected for 18 flavonoids in AMPE2. Each locus could explain phenotypic variation ranging from 6.88 to 19.48%, with a mean of 10.19% (Additional file 4: Table S3). Of the 17 common flavonoids for which significant loci were detected in both experiments, a total of 42 and 27 loci were detected in AMPE1 and AMPE2, respectively, and 12 of which were conserved for the same flavonoids in both experiments (Additional file 5: Figure S2A). The detailed information for GWAS results including P value and R2 of each locus, physical position and minor allele frequency (MAF) of lead SNP and the most likely candidate gene and its annotation are provided in Additional file 4: Table S3. All potential candidate genes and their functional annotations within 100 kb (50 kb upstream and downstream of the lead SNP) of the loci identified from GWAS are listed in Additional file 6: Table S4. For the BB population, 51 and 55 QTL were mapped for 22 flavonoids in BBE1 and BBE2, respectively (Table 2). A total of 99 QTL were detected for the 19 common flavonoids in both experiments (Additional file 7: Table S5), 12 QTL of which were conserved for the same flavonoid in both experiments (Additional file 5: Figure S2B). The percentage of phenotypic variation (R2) that each QTL could explain ranged from 2.94 to 76.79%, with a mean of 10.33% (Additional file 7: Table S5). Twenty-nine QTL that explained greater than 10% of the phenotypic variation (R
2 = 10.03-76.79%) were identified. In the ZY population, a total of 123 QTL were detected in the two experiments (Table 2). Each QTL could explain between 2.85 and 23.17% of phenotypic variation, with an average variation of 9.35%. 47 QTL were identified that explained greater than 10% of the variation (R
2 = 10.02–23.17%). Specifically, 64 QTL were detected for 23 flavonoids in ZYE1 (Table 2), with an R2 range of 4.81 to 23.17% and a mean of 9.38%, while in ZYE2, 59 QTL were identified for 23 flavonoids (Table 2) and an R2 range of 2.85–18.34% with a mean of 9.31% (Additional file 7: Table S5). Of the 21 common flavonoids for which could detected QTL in both experiments, a total of 57 and 51 QTL were detected in ZYE1 and ZYE2, respectively, 27 of which were conserved for the same flavonoids in both experiments (Additional file 5: Figure S2C). Linkage map** results from both BB and ZY populations indicated that most flavonoid QTL were identified with moderate effects (R
2 < 10%), while a relatively small portion showed major effects (27.4% QTL for BB and 38.2% QTL for ZY with an R
2 ≥ 10%). The identified QTL in both RIL populations are evenly distributed across the maize genome, and detailed information for the QTL results, including logarithm of odds (LOD) value, 2-LOD confidence interval, explained phenotypic variation (R2) of each QTL, as well as candidate genes and their annotations are provided in Additional files 7 and 8: Tables S5 and S6. Two and four flavonoids QTL hot spots were observed across the maize genome in the BB and ZY population, respectively, determined by using 500 permutations at the level of 0.05 (Additional file 5: Figure S2B-S2C; Additional file 7: Table S5). These QTLs were shared by flavonoids that are biochemically related and three known flavonoid pathway genes (p1, c2 and mrpa3) located in hot spots on chromosome 1, 4 and 9, respectively (Additional file 5: Figure S2A, S2C). In Additional file 9: Table S7, the co-localization of QTL and/or significant loci identified across different environments or different populations is summarized. Overall, 49 trait-loci combinations that are 25 QTLs corresponding to 23 traits were detected in more than one environments or populations (AMP, BBRIL, ZYRIL) in this study (Additional file 9: Table S7). Among them, 11 combinations (six loci for 11 traits) were detected in more than two environments which including seven combinations (five loci for seven traits) identified in four environments. Detailed analyses of the candidate genes underlying these loci will almost certainly provide useful further information concerning the flavonoid biosynthetic pathway. In our previous study, a primary regulatory network consisted of 58 candidate genes for the flavonoid biosynthetic pathway was constructed using an eQTL and qGWAS method based on the expression level of 15 known maize flavonoid pathway genes [3). In Table 3, we summarized genes for which multiple lines of evidence were provided, i.e., these are genes repeatedly identified in multiple populations or across multiple environments or overlapped genes between the result of network analysis and genetic map** (Table 3). Three of the 11 genes from the primary network and 14 of the 28 genes from the secondary network mentioned above were detected in more than two environments or for more than two flavonoids in one environment, respectively (Table 3). These genes were subsequently prioritized for further functional characterization. 40% of these 45 candidate genes revealed by multiple evidences mentioned above were annotated as enzymes, while functions of 29% of these genes remain unknown. Genes that were annotated as transcription factor and participating in cellular organization only accounted for a small proportion (Additional file 10: Figure S3). According to the map** results and multiple information regarding prior knowledge of flavonoid biosynthesis and functional annotation of candidate genes, we chose several genes that were supported by multiple evidences for further verification. A QTL on chromosome 6 was identified for the level of C-pentosyl-apigenin O-caffeoylhexoside (n1270) in the B73/BY804 RIL population (Fig. 2a). Three genes, GRMZM2G162755 (UGT1, chr6:119876153-119878032), GRMZM2G162783 (UGT3, Chr6:119,862,763-119,864,524) and GRMZM2G383404 (UGT4, chr6:120018887-120020772) which are all annotated as flavonoids 3-O-glucosyltransferase are located within this QTL. UGT1 is about 12Kb upstream of UGT3, and both genes were identified as targets of the R2R3-MYB transcript factor P1 [31]. UGT1 co-expressed with several genes involved in the flavonoid pathway, such as C2 (GRMZM2G422750, chalcone synthase), Chi1 (GRMZM2G155329, chalcone flavanone isomerase 1) and Pr1 (GRMZM2G025832, cytochrome P450) [80]Results
Variation of flavonoids in different maize populations

GWAS for flavonoid levels
Linkage map** for flavonoid levels in the two RIL populations
Candidate genes revealed by multiple evidences
Functional verification of candidate genes underlying the natural variation of flavonoids in the mature maize kernel
Herein we looked into the genetic variations between the two parental lines of the BB RIL population (B73 and By804) and found seven SNPs between the parents in the coding sequence of UGT1, which could cause nonsynonymous mutations (Fig. 2d). Three pairs of KASP (LGC) primers which can successfully genotype three (i.e., SNP811, SNP1331, and SNP1415) of the seven SNPs were used to test the association panel aiming to validate the function of these three SNPs in UGT1. They all exhibited a minor allele frequency (MAF) of more than 0.05. At the sites SNP811 (a Pro to Ala variant) and SNP1331 (a Gly to Glu variant), phenotypic values of lines with the alleles from the two parents were significantly different (t test, P < 0.05; Fig. 2c). Significant phenotypic differences between the lines harboring B73 alleles and By804 alleles were also observed for several other flavonoids detected in this study. For instance, the levels of three apigenin derivatives, chrysoeriol and six chrysoeriol derivatives, four tricin derivatives and cyanidin 3-O-glucoside detected in lines with two parental alleles at SNP811 were significantly different. Similarly, the levels of cyanidin 3-O-glucoside, chrysoeriol di-C-hexoside, 3′,4′,5′-tricetin-O-hexoside and apigenin C-pentosyl-O-coumaroylhexoside in lines with two parental alleles at SNP1331 were significantly different (Additional file 11: Figure S4). In addition, we conducted candidate association analysis using these three SNPs - SNP1331 displayed the lowest P value and can therefore be considered as the most promising functional site among these three SNPs (Additional file 12: Table S8). Compared to SNP811, SNP1331 was associated with more flavonoids, which may suggest that it exhibits broader substrate specificity.
GRMZM2G383404 (UGT4) is around 142 kb away from UGT1, which is associated with the level of apigenin C-pentosyl-C-pentoside (n1201) as revealed by our previous genome wide association analysis, and an amino acid substitution (Asp to Ala) was suggested as one of the functional genetic variants [44]. In the present study, we generated over-expression lines by ectopically expressing GRMZM2G383404 under the control of the maize ubiquitin promoter in the rice cultivar Zhonghua11 (Fig. 3a). We detected the level of flavonoids in the rice leaves of the wild type and T1 individuals of two over-expression lines (L4 and L5) (Fig. 3b). The level of more than half (14/26) of the detected flavonoids were significantly decreased in the over-expression lines. The fold change of these 14 flavonoids between the over-expression lines and wild type ranged from 0.25 to 0.68 (Fig. 3c and Additional file 13: Figure S5). Among them, fold change between the over-expression lines and wild type of the level of apigenin C-pentosyl-C-pentoside was around 0.65. Along with apigenin C-pentosyl-C-pentoside, two other apigenin derivatives (i.e., apigenin 7-O-glucoside and apigenin di-C-hexoside) were also affected (Additional file 13: Figure S5). Notably, the level of all the tricin derivatives detected here (and tricin itself) was significantly decreased (Additional file 13: Figure S5). Moreover, the content of chrysoeriol, chrysoeriol O-hexoside and vitexin were also significantly decreased (Additional file 13: Figure S5). However, no significant changes were found for the content of C-pentosyl-apigenin O-caffeoylhexoside, for which UGT4 was identified in the QTL region as mentioned above. Hence, UGT1 and UGT3 but not UGT4 could be the causative genes for the variance of C-pentosyl-apigenin O-caffeoylhexoside. However, the transgenic result of UGT4 can suggest its influence in the flavonoid biosynthesis. However, further biochemical assay is needed to strongly confirm the function and activity.

Transgenic result of UGT4. a Diagram of over-expression construct. b The bar plot showing the average mRNA level of UGT4 (GRMZM2G383404) in the wild type (WT) and over-expression lines (T1) (the individual number is 9, 5, 9 for WT, L4 and L5, respectively, 3 technical replicates for each line). c The bar plot showing the relative contents of apigenin C-pentosyl-C-pentoside (n1201) in the WT and UGT4 over-expression lines (n = 9, 5, 9 for WT, L4 and L5, respectively), * and ** represent the significant level of P < 0.05 and P 0.01, respectively
On chromosome 2, gene GRMZM5G843555 was suggested to be important in determining the level of apigenin C-pentosyl-O-coumaroyl hexoside (n1268) by both linkage map** in Zong3/Yu87-1 population (Fig. 4a) and GWAS in AMPE1 (Fig. 4c). GRMZM5G843555 is annotated as an oxoglutarate/iron-dependent oxygenase (OXY), which belongs to the oxygenase superfamily. However, GRMZM5G843555 (OXY) shows low sequence similarity with the well-known 2-ODD genes, such as FNS. OXY is one of the maize prolyl 4-hydroxylase family (P4Hs) members, which may play a role in tolerance to abiotic stresses, such as water-logging [48]. Correlations between the content of various flavonoids and the expression level of OXY revealed that the content of chrysoeriol, chrysoeriol O-rhamnosyl-O-hexoside, tricin O-rhamnosyl-O-hexoside and 3′,4′,5′-tricetin O-rhamnosyl-O-hexoside, chrysoeriol O-hexoside, chrysoeriol di-C-hexoside and chrysoeriol C-hexosyl-O-rhamnoside were negatively correlated with expression level of OXY (r = -0.19 ~ -0.1; p < 0.05) (Fig. 4d). We further profiled the rice over-expression lines and quantified the level of 26 flavonoids (Fig. 4b). The levels of 20 flavonoids were significantly decreased compared with that of the wild type (Fig. 4e-h, Additional file 14: Figure S6). Within these 20 flavonoids, the content of six flavonoids was negatively correlated with the OXY expression level. Based on the result, we speculate that the OXY may act as a competitor or inhibitor of the flux through the apigenin, chrysoeriol and tricin branches of flavonoid metabolism.

Linkage and association map** of OXY and validation by transformation. a and c Diagram of linkage map** and GWAS results for the level of Apigenin C-pentosyl-O-coumaroylhexoside in maize kernel. LOD values are shown as a function of their genetic positions. And the peak SNP is located within OXY (GRMZM5G843555). b The bar plot showing the mRNA level of OXY in wild type (WT) and over-expression lines (T1) (the individual number is 6, 10, 7, 8 for WT, L31, L35, L36 and L37, respectively, 3 technical replicates for each line individual). d Plot of correlation between the content of different flavonoids and the normalized expression level of gene OXY in association panel. e-h The bar plot for the relative contents (fold change relative to the mean level of each flavonoid) of naringenin, chrysoeriol, vitexin and tricin between the WT and OXY over-expression lines (n = 6, 10, 10, 10 respectively). * and ** represent the significant level of P < 0.05 and P < 0.01, respectively
In addition, abundant genetic variants between Zong3 and Yu87-1 in the promoter region of OXY were observed (Additional file 15: Figure S7). Cis-element prediction using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) found a variant between the sequences of the two parental lines at the MBS II (MYB binding site II, [49]). The variant at this binding site may affect the function of OXY through transcriptional regulation, as suggested by the finding in Petunia Hybrida [49]. Indeed, a strong cis-eQTL for OXY was identified in our previous study, which may suggest the potential function of this genetic variant at the upstream of this gene (Additional file 16: Figure S8).
To investigate the co-expression mode of the above mentioned genes, a qGWAS-based network was constructed (Fig. 5). GRMZM5G843555 (OXY) is not in this network for no related genes found by using the threshold of P < 3.5 × 10−7 (0.01/28369). UGT4 clustered independently from the rest. Four well-known genes involving in the flavonoids biosynthesis are present in the co-expression network, such as a1 (GRMZM2G026930), c2 (GRMZM2G422750), chi1 (GRMZM2G155329) and whp1 (GRMZM2G151227). 22 uncharacterized genes are also revealed in the network, including a gene homologous to chalcone isomerase (GRMZM2G175076) and other 21 genes with unknown function or without direct functional annotations related to flavonoid biosynthesis.

Co-expression network for UGT1, UGT2, UGT3 and UGT4 based on a qGWAS method. The red indicates these four genes. The green diamond indicates the known enzyme involved in the flavonoid pathway. The blue circle indicates the uncharacterized co-expressing genes
Discussion
Metabolomics, which promotes the study of plant metabolism, offers the capacity to speed up the breeding process toward high yielding and nutritional crops [50, 51]. With the advent of high-efficiency metabolic profiling and high-throughput sequencing technologies, studies of genetic dissection of metabolomics diversity based on GWAS and linkage analysis have been reported recently in several plant organisms such as maize [44, 52], rice [41, 57] and Arabidopsis thaliana [58, 59]. In addition to the dataset generated from our previous untargeted metabolomics-based genetic map**, here we focus on the flavonoids that were found in the mature kernel harvested from an association panel and two RIL populations grown across multiple environments [44, 2b), are located in one glycosylated flavone QTL region (Fig. 2a). The function of UGT3 was elucidated as having both C- and O- glucosyltransferase ability [29], while the biochemical activity of UGT1 and UGT4 remains unknown. Re-sequencing the two parental lines as well as the whole association panel and subsequent candidate association analysis have provided us with the potential functional genetic variants of UGT1. However, whether and if so how the two amino acid replacements in the coding region of UGT1 influence the protein structure and enzymatic activity remains to be answered. In addition given the genomic location of these three UGT genes, it will also be interesting to look into the genomic divergence of the region covering the UGT1 locus between diverse maize germplasm (including inbred lines, landraces and wild progenitors) from an evolutionary perspective. On the other hand, we found significant decreases of several flavonoids in UGT4 overexpressing rice leaves. And in our co-expression network, UGT1, UGT2 and UGT3 are clustered with four well-characterized flavonoids biosynthesis pathway genes, while UGT4 clustered independently from the rest. Further molecular and biochemical evidence will be needed to provide the exact mechanism underlying this observation. In addition, the maize OXY gene exerted a negative influence on the levels of a broad range of flavones in the over-expressing rice leaves.
It has been documented that flavonoids have potent bioactivities which are beneficial for human health. Epidemiologic studies have suggested that a diet rich in flavones exhibits some anti-carcinogenic as well as anti-angiogenic and anti-inflammatory bioactivities [60–62]. Flavonoids also have antioxidant and anti-diabetic potential when added to food [12] as a means to probe the activity of metabolic enzymes in an approach that circumvents the need for transgenic plants. This method as well as the approach we took in the current study offers new opportunities to advance beyond the QTL/association map** approach and towards a complete understanding of maize flavonoid biosynthesis.
Methods
Plant materials, genotypic and metabolic data
The metabolic data used in this study is measured from genetic materials including an association map** panel (referred to as AMP hereafter) for GWAS and two Recombinant Inbred Lines populations (RILs; BB and ZY) for linkage analysis as described previously [44, 70] and linkage map was constructed using recombinant bins for both RIL populations. Briefly, a map containing 2,496 and 3,071 unique bins was constructed for BB and ZY RILs, respectively [71].
Genetic map**
Genome-wide association study (GWAS) was performed using a compressed mixed linear model (cMLM) implemented in the software TASSEL 3.0, accounting for the population structure (Q) and familial relationship (K) [72]. SNPs with minor allele frequency (MAF ≥ 5%) in the 368 lines were employed in the association analysis. To facilitate the interpretation of GWAS results, P value of each SNP was calculated and significance was defined at a uniform threshold of 1.8 × 10−6 (i.e., P ≤ 1/N, N = 556809 which is roughly a Bonferroni correction). SNP with the lowest P value (i.e., the lead SNP) and its corresponding gene were reported for each significant flavonoid locus (see Additional file 4: Table S3). Linkage map** was conducted using composite interval map** method [73] implemented in Windows QTL Cartographer V2.5 for each flavonoid trait identified in both RIL populations [74]. Zmap (model 6) with a 10-cM window and a walking speed of 0.5 cM was used. To determine a threshold for significant QTLs, 500 permutations (P = 0.05) were used for each flavonoid identified in both RIL populations. The bins were clearly defined, and a uniform LOD value was assigned for each bin. The confidence interval for each QTL was assigned as a 2 LOD drop from the peak. The setting of parameters was the same as described previously [47]. Detailed information including physical location, confidence interval, and R 2 (explained phenotypic variance) of each QTL for each flavonoid trait is shown in Additional file 7: Table S5. To test the cross-validation between GWAS and linkage analysis, 200 kb region of loci identified by GWAS (the 100 kb upstream and downstream region of the lead SNP) was compared with the physical region of QTL.
Candidate gene identification
The filtered working gene list of maize genome was downloaded from MaizeGDB (http://www.maizegdb.org) to identify possible candidate genes in each QTL. Candidate genes were annotated according to InterProScan (http://www.ebi.ac.uk/interpro/scan.html). All potential candidate genes and their annotations within 100 kb (50 kb upstream and downstream of the lead SNP) of the loci identified from GWAS are listed in Additional file 6: Table S4. Candidate genes associated with the corresponding flavonoid trait that were searched within the confidence interval for each QTL from linkage map** are listed in Additional file 8: Table S6. The most likely candidate gene was selected by testing for either gene flavonoid association or association between the gene and pathway. For the loci without appropriate candidates, the gene nearest to the lead SNP is assigned.
Constructs and transformation
To generate OXY over-expressing constructs, the vector pCAMBIA1301s which contains the selectable marker gene hpt and 35S promoter was used. There are six transcripts for OXY on the B73 reference genome. To decide which one to be transferred, five genotypes with highest n1268 content and five with lowest n1268 content were chosen to compare the abundance between different transcripts. We observed that transcript T04 with six exons was the most highly expressed. So the B73 genomic DNA fragment of OXY T04 (from the ATG to TGA) was amplified and the PCR product was cloned into vector pCAMBIA1301s with restriction enzyme KpnI and XbaI. On the other hand, gene UGT4 has only one transcript on B73 genome. Therefore UGT4 T01 DNA fragment of B73 (from ATG to TGA) was amplified to generate the UGT4 over-expressing construct into vector pCAMBIA 1300nu (with the selectable marker gene hpt) using restriction enzyme BamHI and KpnI. The final plant expression vector was introduced into Agrobacterium EHA105 by electroporation and calli induced from mature seeds of an elite japonica rice cultivar Zhonghua11 were used for Agrobacterium-mediated transformation [75].
Expression analyses
We isolated total RNA from rice leaves with TRIzol (Invitrogen) as the manufacturer’s instructions. The first-strand cDNA was synthesis using a TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TansGen Bioteke) according to the manufactures’ protocol. Quantitative PCR was performed on an optical 96-well plate in a BioRAD PCR system (CFX96) with SYBR Mix (Vazyme). The relative expression level of gene OXY and UGT4 was determined with the rice Actin as an internal control. The expression measurements were obtained using the relative quantification method [76]. The leaves of transgenic rice are sampled and stored immediately in liquid nitrogen. The following extraction and flavonoid profiling were as described previously [44]. A student’s t-test was applied to examine the difference between over-expression line and the wild line.
Phylogenetic analysis
UGT protein sequences were aligned using CLUSTAL W implemented in MEGA7 (version 7, http://www.megasoftware.net/) [77]. A phylogenetic tree was constructed from aligned UGT protein sequences by MEGA7 using the neighbor-joining method [78] with the following parameters: bootstrap method (1000 replicates), Poisson model, uniform rates, and complete deletion.
Re-sequencing and allele identification
For UGT1 (GRMZM2G162755) and OXY (GRMZM5G843555), the PCR fragments from gDNA of B73 and By804 were sequenced via the Sanger re-sequence method. The SNPs were identified using CLUSTAL OMEGA online (http://www.ebi.ac.uk/Tools/msa/clustalo/). We designed one of the two direction primers with 3′end at the SNPs of UGT1 according to the direction provided by LGC (Laboratory of the Government Chemist) and performed PCR with the KASP Assay Mix referring to its procedure. All primers for vector construction and re-sequencing used in this study are shown in Additional file 17: Table S9.
Construction of co-expression network
A qGWAS-based method was adopted to construct the co-expression network as described previously [47]. The expression data was obtained from our previous RNA sequencing analysis on maize kernels (at the stage of 15 day after pollination) of the association panel containing 368 maize inbred lines [68]. We focused on the four candidate genes (UGT1, UGT2, UGT3, and UGT4) and their co-expressing genes with the threshold of P < 3.5 × 10−7(0.01/28369). The program Cytoscape [79] was used to display the network.
