
Abstract
Background
Belonging to the Actinobacteria phylum, members of the Rhodococcus genus thrive in soil, water, and even intracellularly. While most species are non-pathogenic, several cause respiratory disease in animals and, more rarely, in humans. Over 100 phages that infect Rhodococcus species have been isolated but despite their importance for Rhodococcus ecology and biotechnology applications, little is known regarding the molecular genetic interactions between phage and host during infection. To address this need, we report RNA-Seq analysis of a novel Rhodococcus erythopolis phage, WC1, analyzing both the phage and host transcriptome at various stages throughout the infection process.
Results
By five minutes post-infection WC1 showed upregulation of a CAS-4 family exonuclease, putative immunity repressor, an anti-restriction protein, while the host showed strong upregulation of DNA replication, SOS repair, and ribosomal protein genes. By 30 min post-infection, WC1 DNA synthesis genes were strongly upregulated while the host showed increased expression of transcriptional and translational machinery and downregulation of genes involved in carbon, energy, and lipid metabolism pathways. By 60 min WC1 strongly upregulated structural genes while the host showed a dramatic disruption of metal ion homeostasis. There was significant expression of both host and phage non-coding genes at all time points. While host gene expression declined over the course of infection, our results indicate that phage may exert more selective control, preserving the host’s regulatory mechanisms to create an environment conducive for virion production.
Conclusions
The Rhodococcus genus is well recognized for its ability to synthesize valuable compounds, particularly steroids, as well as its capacity to degrade a wide range of harmful environmental pollutants. A detailed understanding of these phage-host interactions and gene expression is not only essential for understanding the ecology of this important genus, but will also facilitate development of phage-mediated strategies for bioremediation as well as biocontrol in industrial processes and biomedical applications. Given the current lack of detailed global gene expression studies on any Rhodococcus species, our study addresses a pressing need to identify tools and genes, such as F6 and rpf, that can enhance the capacity of Rhodococcus species for bioremediation, biosynthesis and pathogen control.
Similar content being viewed by others

Background
Rhodococcus species comprise a genus of gram-positive actinomycetes in the Actinobacteria phylum best known for their ability to metabolize environmental pollutants and the capability to produce desirable biological compounds such as steroids [1,2,3,4,5]. Due to a high tolerance to toxic substrates, the capacity to degrade a wide range of organic and xenobiotic substances, and the ability to produce biosurfactant, Rhodococcus spp. are ideal candidates for the bioremediation of contaminated sites [3]. Members of the genus have a broad geographic range, and have been isolated from a variety of environments, most often soil including at contaminated sites, but also from freshwater, wastewater, sediment, air, and crude oil [2, 6,7,8]. They have also been found in host-associated systems, and while most species are benign, some can cause infections in plants [9] and animals [10], and more rarely in humans [11, 12].
Bacteriophage, viruses which infect bacteria, are the most abundant biological entities on earth, and are generally considered to be ten times as numerous as their hosts in any environment [13]. Dynamics in natural ecosystems are mediated by phage-host interactions, and phage predation has the potential to be exploited for use in industrial and biomedical applications. R. opacus phage Toil has been used as a bioextraction agent for biodiesel production, forcing the release of triacylglycerols from its host upon lytic infection [14]. Phage of Rhodococcus and other related Actinobacteria isolated from wastewater have been suggested for use as biocontrol agents to reduce foaming in activated sludge [15, 16]. Phage YF1, which can infect R. equi, R. erythropolis, R. rhodochrous, and R. opacus, was used to identify potential new antimicrobial targets in its hosts, serving as a model for novel target discovery in other related organisms [17].
A significant number of Rhodococcal phage have been isolated and sequenced; NCBI and the Actinobacteria Phage Database currently contain entries for 74 sequenced phage of 4 Rhodococcus species [18]. The most numerous of these are phage that infect Rhodococcus erythropolis RIA-643, many of which have been characterized in conjunction with the Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) program [19, 20]. The majority of these sequenced phage have been classified in cluster CA, a group of temperate phage which share a common genomic architecture [20, 21]. Cluster CA phage have regulatory structures and a large subset of protein coding genes similar to cluster A mycobacteriophages [20]. Phage gene expression during infection and, in some cases, corresponding host responses have been described at the molecular level for mycobacteriophage [22,23,24]. However, despite their importance, exploration of transcriptional dynamics remains largely unexplored for Rhodococcus phage and their hosts.
Here, we present Winter Compost 1 (WC1), a new phage of Rhodococcus erythropolis RIA-643 isolated in Williamsburg, Virginia in 2018. We characterize WC1 at the genomic level, and analyze temporal transcriptional profiles of both phage and host during infection using RNA-seq. The genome of WC1 was typical of cluster CA phage with high (> 90%) levels of nucleotide similarity and between coding sequences. During host infection WC1 genes were expressed following a temporal program of early, middle, and late, which was also mirrored by the spatial organization of transcribed regions in the genome. WC1 infection results in a dramatic suppression of host metabolism and a notable disruption of metal ion, particularly, iron homeostasis. By 120 min, 80% of the transcripts map to phage genes. Only 13% of host genes are differentially expressed. This work provides insight into how a cluster CA phage alters the global transcription program of its host during infection, and may apply more broadly to other Rhodococcal phage-host systems. A more detailed understanding of these phage-host interactions can facilitate development of phage-mediated strategies for bioremediation as well as biocontrol in industrial processes and biomedical applications.
Methods
Rhodococcus erythropolis host strain and WC1 bacteriophage isolation
Rhodococcus erythropolis RIA-643 [BUCSAV 57.1] was obtained from ATCC (ATCC® 15,903™). Cells were initially grown in Middlebrook 7H9 media with AD supplement (10%), carbenicillin (50 µg/ml), cyclohexamide (10 µg/ml), and calcium chloride (1 mM). For RNA-Seq experiments carbenicillin and cyclohexamide were omitted. Winter Compost 1 Phage (WC1) was isolated from a compost sample at 37.282925 N, 76.664688 W in 2014 using a standard enrichment protocol [18, 23]. Briefly the soil sample was incubated with a culture of Rhodococcus erythropolis in media for 24 h at 37 C. The sample was filtered with a 0.2 μm PES filter and 100 µl of the filtrate and 500 µl of a R. erythropolis culture was combined with 7H9 top agar and plated on LB plates. Plaques were visible within 24 h; plaques were subjected to three rounds of purification to ensure a plaque pure sample.
WC1 bacteriophage DNA isolation and sequencing
Following plaque purification, high titer lysate was obtained by flooding host plates displaying web lysis with 1X phage buffer (10mM Tris, pH 7.5, 10 mM MgSO4, and 0.4% w/v NaCl). To isolate phage DNA, the lysate was treated with nuclease mix (0.8 U/ml DNase I and 100 µg/ml RNase) with 12.5mM MgCl2 at 37 °C for 30 min in order to remove bacterial DNA. Phage capsid was digested using proteinase K (500 µg/mL) and a detergent (0.5% SDS) after stop** DNase activity using a bivalent cation chelator (2mM EDTA) at 55 °C for 60 min. Phage genomic DNA was extracted using Phenol:Chloroform:Isoamyl alcohol (25:24:1), and precipitated using sodium acetate (0.3 M) and ice cold ethanol. Finally, the DNA was collected by centrifuging 13,000 rpm for 10 min; the DNA pellet was washed with 70% ethanol and re-suspended in nuclease free water. Phage genomic DNA was sequenced using the Ion Torrent PGM Sequencer system with standard library preparation according to the manufacturer’s protocol for 150 bp reads (Ion PGM Sequencing Kit, 314 Chip v2).
WC1 genome assembly and annotation
Raw WC1 reads were quality checked using FastQC v0.11.5 [25]. Reads were trimmed using Trimmomatic v3.6 [26] with default parameters resulting in 1.2 million reads, giving 7500X coverage of the genome. De novo genome assembly was performed using the CLC Microbial Genomics Module to obtain major contigs with a minimum of 100X coverage, and finishing was performed with Consed [27] and the CLC Microbial Genomics Module (Qiagen) according to the protocols provided at [28]. Initial gene prediction (features) were obtained using DNA Master [29], which makes use of Glimmer [30], GeneMarkS [31], and Aragorn (Laslett and Canback 2004). In the process of gene-by-gene refinement, the features were then manually modified, deleted, or inserted following the Guiding Principles of Bacteriophage Genome Annotation (06/16/2018) available via SEA PHAGES [19]. Decisions were made based on coding potential (GeneMark Version 2.5p), RBS score (using SD Scoring Matrix Kibler6 and Spacing Weight Matrix Karlin Medium available in DNA Master), length of ORF, number of gaps or overlap** nucleotides, and BLASTp match (evalue < 10− 4). The tRNA genes predicted by DNA Master were further confirmed using ARAGORN v1.2.38 [32] and tRNAscan-SE v. 2.0 [33]. For protein assignments, each gene was evaluated using HHPred [34] and BLASTp [35]. Context of the functions of the flanking genes (synteny) was examined using Phamerator [36].
Phage comparative genomics
Genomes for 38 previously isolated phage of R. erythopolis RIA-643 were obtained from Genbank (accession numbers and cluster IDs appear in Table S1). This includes 34 phages from Cluster CA (temperate), 3 from cluster CB (lytic), and 1 from cluster CE (unknown lifestyle) of Rhodococcus phage as delineated by phagesdb [18]. GC content and pairwise nucleotide similarity between WC1 and all genomes was determined using the online tool JSpecies WS [37] with the ANIM MUMmer-based option. Genome architectures were obtained from Phamerator [36] for comparison with WC1.
One-step growth curve experiment
R. erythropolis RIA-643 log phase liquid-culture and WC1 bacteriophage were combined at a multiplicity of infection of 1:10. Cells were incubated at 37 °C for 5 min to allow phage adsorption, and then centrifuged for 3 min at 5,000G at 4 °C. Pelleted cells were washed to remove unadsorbed phage particles using 1 mL room temperature phage buffer (10mMTris pH 7.5, 10mMMgSO4, 4% w/v NaCl). Cells were subjected to three rounds of washing, each of which was followed by centrifugation at 5,000G for 3 min at 4 °C. Final bacterial pellets were then resuspended in 40 ml Middlebrook 7H9 media. For the duration of the experiment, cells were incubated in with shaking 37 °C. 200 µL samples were taken at 5,
30, 36, 50, and 60 min and then every subsequent 30 min over a 4 h period and were serially diluted in phage buffer. Samples were then added to 500 µL of uninfected R. erythropolis, incubated for 5 min at room temperature, and plated for measurement of viral titer to assess the length of the latent period.
RNA preparation and sequencing
To obtain RNA samples, 15 mL of R. erythropolis cell were collected prior to addition of phage. For the 5, 30, 60, and 120 min time points, 15 mL of phage-infected R. erythropolis cells were. Cells were centrifuged at 4000 G at 4 °C for 10 min. The resulting pellets were flash frozen in liquid nitrogen and transferred to -80 °C for storage. One ml of TRIzol (Life Technologies) was used to resuspend frozen pelleted cells, and total RNA was extracted as previously described [23]. The RiboZero kit for gram-positive bacteria (Illumina) was used to deplete ribosomal RNA from RNA extraction samples following manufacturer’s instructions. Following depletion, RNA library size and effectiveness of rRNA depletion was checked using a Bioanalyzer 2100 (Agilent). rRNA-depleted RNA was then prepared for sequencing using the TruSeq Stranded RNA-Seq Kit (Illumina) and sequenced on an Illumina Miseq (150 bp reads) according to the manufacturer’s instructions generating a total of 55,302,168 paired-end reads. All reads used for this study had a Q35 score. Raw RNA-Seq reads can be accessed via NCBI’s Short Read Archive at project accession number SRP154435.
Differential expression analysis
Sequence length, quality, and composition for raw reverse-stranded RNA-Seq reads which contained both bacteria and bacteriophage transcripts evaluated using FastQC v0.20.0 [25]. Trimming and filtering was performed using Trimmomatic v3.9 [26] with the parameters SLIDINGWINDOW:4:15 TRAILING:20 MINLEN:75 AVGQUAL:20 based on review of the FastQC outputs. Libraries were aligned to phage and host genomes using HISAT2 v2.2.1 [38] excluding multi-map** reads. Read coverage was assessed using the bamtools coverage tool [39], with subsequent read counts normalized by library size and averaged over replicates. The resulting alignments were mapped to features for the phage and host genomes using FeatureCounts [40] from the SubRead Anaconda package version 2.0.3, with option -s 2 to only assign reads if they align reverse-stranded to a feature, -p to count paired end reads as a single fragment, and the options -O and --fraction to count overlap** reads and assign fractional amounts of reads according to the number of features that a read overlaps.
Differential expression (DE) analysis was conducted using the R package DESeq2 (version 1.38.3) [41], using ashr for LFC shrinkage [42]. Pairwise comparisons between all time points were conducted using individual Wald tests. Results were filtered to adjusted p-value of 0.05 for Wald tests, as well as minimum absolute log2 fold change of approximately 0.58, representing a minimum absolute fold change of 1.5, which is in accordance with guidelines established by Schurch et al. [43].Hierarchical clustering of phage genes was performed using scikit-learn AgglomerativeClustering with complete linkage. The optimal number of clusters was determined by creating a dendrogram containing the full tree.
Host RNA-seq functional enrichment analysis
GO terms were assigned to R. erythopolis RIA-643 genes using a combination of bioinformatic strategies which allowed for cross-referencing and confirmation of term assignments. Automated analyses were carried out using eggNOG-mapper (http://eggnog-mapper.embl.de/) [44], and the command line version of InterProScan v5.63-95 (https://www.ebi.ac.uk/interpro/about/interproscan/) [45, 46]. GO terms were also assigned using a modification of the approach available at https://github.com/enormandeau/go_enrichment. Specifically, all bacterial coding sequences were translated using gffread [47] and compared to the full Swissprot [48] database (available from: ftp://ftp.ncbi.nlm.nih.gov/blast/db/swissprot) using BLASTp (e-value < 0.001, max of 1 target sequence) [35]. BLAST hits were filtered using the parameters outlined in Rost (1999) which use require an adjusted cutoff percent similarity based on the length of residues aligned. Annotations for significant hits were retrieved from UniProtKB [48]via the RESTful API and were parsed for GO terms. Terms assigned to genes by more than one method were cross-validated, and the union of all assignments was used as the final set of GO terms, resulting in 4846 annotated coding sequences. All annotated bacterial coding sequences were functionally annotated with KEGG terms using the KEGG Orthology Based Annotation System Intelligent version (KOBAS-i) . The annotation program was run using the closest KEGG annotated reference organism, Rhodococcus sp.008, and resulted in 6233 annotated and 602 unannotated coding sequences. Enrichment analysis for GO and KEGG terms was performed in R with ClusterProfiler (version 4.6.2) on log2 fold-change values for all genes that were not filtered by DESeq2 independent filtering. The enricher function was used for GO, and the enrichKEGG function for KEGG. Non-coding RNAs found to be DE in the host were characterized using the Rfam database [49]. GO terms were condensed based on term similarity using the command line tool GO-Fig. [50].
Results
WC1 genome architecture is concordant with other temperate R. erythropolis phage
The WC1 genome contained 46,439 base pairs, with a GC content of 58.6%. The length and GC content were consistent with cluster CA phage of R. erythropolis RIA-643, which are known to be temperate (Figure S1) [20]. WC1 had high average nucleotide identity to other R. erythropolis cluster CA phages, ranging from a low of 92.7% (Partridge) to a high of 98.6% (Erik). Within the WC1 genome, 66 protein coding genes were identified, of which, 35 (53%) were annotated with putative functions (Figure S2). Three tRNAs were annotated on the far left arm of the genome, approximately 1.5 kb from the origin, as seen previously in cluster CA phage [20].
Overall, the genomic content and architecture of WC1 shows markedly high synteny with previously described temperate phages of R. erythropolis RIA-643. As described in [20], this includes, from left to right, the three tRNAs previously mentioned, a cluster of genes related to virion structure and assembly, a putative immunity repressor, and a genetic cluster with functions necessary for replication and regulation (Figure S3). There are 16 copies (13 forward, 3 reverse complement) of the 13mer 5’-YGWCTATTGTCAA-3’ primarily (75%) in intergenic regions of WC1 (Figure S2). These regions have previously been described in R. erythropolis cluster CA phage as well as cluster A mycobacteriophage, and have a putative regulatory function, maintaining lysogeny by binding the immunity repressor [20, 51].
Phage transcripts dominate during the infection time course
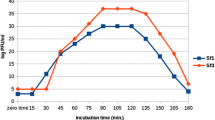
Dual RNA-sequencing of the host and the infecting phage was performed prior to infection (0 min) and at four timepoints up to 120 min after infection. Time points for RNA-seq were chosen based on exploration of the phage lifecycle via a one-step growth experiment (Fig. 1A). Specifically, the 5 and 30 min time points represent early and late phases in the latent period respectively; the 60 min time point corresponds to the lysis phase, and the 120 min time point lies at the point of what appears to be a second burst, indicative of another round of infection (Fig. 1A).

A: Phage titer during host infection. Grey lines and circles represent individual replicate experiments. The blue line shows the average. B: Percentage of reads at each time point mapped to host versus phage genomes. Solid circles show averages of two replicates and the x labels show individual replicates – in most cases these are close to the mean and thus do not appear
Overall, for all replicates at all time points, the proportion of total RNA-Seq reads mapped to either the phage or host genome ranged from 94.72 to 97.92% (Table S2). Over the time course of the infection, the proportion of total RNA-Seq reads aligning to the host showed a marked decrease while reads aligning to the phage genome showed the opposite trend, increasing to approximately 81% of total aligned reads at 120 min (Fig. 1B). A small number of reads aligned to phage features at time 0, but these represented less than 0.02% of total reads aligned (Table S3).
Transcriptional activation of phage genes follows temporal and spatial patterns
Phage genes demonstrated temporal expression patterns which largely corresponded to specific regions of the phage genome (Fig. 2). The 69 phage genes were grouped using hierarchical clustering of normalized log counts (see Methods), which were indicative of three distinct temporal patterns: early, middle, and late stage genes. The stage of each gene corresponds in general to its peak expression. Genes in the early, middle, and late classes tended to be spatially clustered near each other in the genome (Fig. 2B). A fourth cluster contained 5 genes with relatively lower expression across all timepoints: 3 corresponding to proteins of unknown function, and the phage serine integrase, and excise genes (Fig. 2A).

Temporal and spatial patterns of transcriptional activation in WC1. (A) Phage expression patterns over time. Parallel coordinate plots (left) show relative counts of phage transcripts for Early, Middle, and Late genes. The counts are transcripts per million (TPM) normalized internally for each gene, i.e. with respect to the lowest expression level for that individual gene. The heatmap (right) shows log TPM averaged at each time point and subsequently standardized. The standardization is performed by column. (B) Genomic locations of early, middle, and late genes in WC1. Colors used in the genome map correspond to those in A
Five minutes post-infection, the most highly expressed genes were localized to the right arm of the phage genome (Fig. 2). Early genes demonstrated high expression levels five minutes after infection, which were sustained or minimally decreased at 30 min, and then subsequently declined (Fig. 2). WC1_68 (a hypothetical protein with unknown function) was the most highly expressed transcript at the five minute time point. The majority of these genes corresponded to proteins of unknown function. The cluster also included a Cas exonuclease, an immunity repressor, and an anti-restriction protein.
At 30 min post-infection, the majority of phage genes exhibited high levels of expression. Middle genes specifically reached peak expression at the 30 min time point and represented genes to the left of the early cluster (Fig. 2). This group contained several genes related to DNA synthesis and replication: deoxycytidylate deaminase, ThyX thymidylate synthase, ribonucleotide reductase phosphoesterase, NrdH glutaredoxin, and a dsDNA helicase. Genes involved in phage genome replication were also highly expressed, including DNA polymerase I, two DNA primases, a DNA-binding protein, and a `phage membrane protein. The other 10 genes in the middle cluster encoded proteins of unknown function.
By 60 min post-infection, the left arm of the phage genome demonstrated markedly higher levels of expression (Fig. 2). The late stage cluster demonstrated peak expression at 60 min, which was sustained for nearly all genes at 120 min, while the genes in the other clusters showed decreased expression at both 60 and 120 min. The late genes were comprised largely of phage structural proteins, phage assembly and packaging proteins, and host lysis and degradation proteins including an endolysin and a holin. The remaining five protein-encoding genes in this cluster included the terminase, both the small and large subunits, and several which had unknown functions; the terminase, small subunit was the most highly expressed genome feature at both 60 and 120 min post-infection. Three phage-encoded tRNAs also clustered with the late group due to sustained high expression at 60 and 120, but displayed similarly high expression at 30 min.
Differential expression (DE) analysis further reinforced these patterns as well as the dominance of phage transcripts during infection (Fig. 3). By 5 min post-infection, 56 phage genes showed significant (Wald test, p-value < 0.05, FC > 1.5) increases in expression when compared to the 0 min baseline (Fig. 3A). The 13 genes that were not significant belonged to the left arm of the WC-1 genome, specifically WC-1 genes 2–7, 13, 19,20, 22, and 25–26, with the single exception of WC1_69. By 30 min post-infection, all 69 phage genes were up-regulated as compared to baseline, and this was sustained at 60 and 120 min (Fig. 3B-D). By 120 min post-infection all phage genes had log2 fold changes greater than 1.5 and p-values less than 0.00001. Sequential comparisons over the time course of infection mirror sequential activation of phage genomic regions (Figure S4; Figure S5).

Differentially expressed (DE) genes over the time course of infection. A-D: Volcano plots for each time point compared to baseline. Each point on the volcano plot is based on 2 replicates. Vertical lines indicate Log2 Fold Change of 0.58 and − 0.58, which corresponds to a fold change of 1.5. The horizontal line indicates p of 0.05. E: Counts of up and down-regulated host genes as compared to the 0 min baseline. Genes considered DE had FC greater than 1.5 and p-values less than 0.05
Non-coding regions of WC-1 were the most highly transcribed genomic loci post-infection
Phage WC-1 contains three transcribed intergenic non-coding regions: between WC1_1 and WC1_2 (nc_1), between WC1_29 and WC1_30 (nc_2), and between WC1_68 and WC1_69 (nc_3). All three regions were most highly expressed 120 min after infection relative to other time points (Fig. 4). At 120 min as well as at 30 and 60 min post-infection, these regions had the highest relative read coverage when compared to the rest of the genome (Figure S6). Of these, region nc_1 had the highest normalized mapped read counts at 30, 60, and 120 min relative to other time points (Fig. 4A), but also relative to all other genome features (Figure S6). This region was not active at 5 min post-infection, which is in concordance with gene expression on the left arm of the genome, which was most active at later in the time course of infection. Region nc_2 (Fig. 4B; Figure S6) showed minimal transcription at 5 min, corresponding to low activation in the central region of the genome overall. In contrast, region nc_3 showed high levels of transcription at 5 min, as did all features in the right arm of the genome (Fig. 4C; Figure S6).

Transcribed intergenic non-coding regions in WC1. The y axis shows reads normalized per million aligned transcripts in each individual library and then averaged for each set of replicates at each genomic location
A core set of host genes were differentially expressed throughout infection
Of the 6928 identified genes of RIA-643, a total of 874 unique host transcripts were differentially expressed over the time course of the infection when each time point was compared to the baseline (Fig. 3). Overall, the proportion of up- versus down-regulated host genes, whether DE or not, were nearly equal, with slightly more suppression than activation at all time points (Fig. 3E). The number of host DE genes varied between time points (Fig. 3), with slightly more up-regulated DE genes at 5 and 120 min, and slightly more down-regulated at 30 and 60 min (Fig. 3E). A core set of 201 genes and 2 non-coding RNAs were DE at every time point with fold change greater than 1.5 as compared to time 0 (Figures S7, S8, S9; Table S4). Of these, 37 encoded proteins of unknown functions.
Two non-coding RNAs were up-regulated at all timepoints versus the baseline (Figure S8). During the course of phage infection in RIA-643, the small RNA F6 (DVG80_03131) reached peak expression at 30 min with lower expression levels at 60 and 120 min. The F6 small RNA (sRNA) in RIA-643 shares 100% identity with 8 other identified F6 sRNAs in the Rfam database, which are found in other strains of R. erthryopolis and other Rhodococcus species. This sRNA is part of Rfam family F6 (RF01791). F6 has been shown to have a regulatory function in Mycobacterium species, associated with and up-regulated during stress responses [52,53,54]. The ydaO/yuaA leader (DVG80_05852) sequence was most highly expressed at 120 min versus the baseline. This RNA was identical to the ydaO/yuaA leader sequence in R. erythropolis PR4, and is part of the ydaO-yuaA (RF00379) Rfam family. ydaO-yuaA is a riboswitch that responds to cyclic di-AMP, serving a regulatory role in transcription [55, 56].
There were 85 universally up-regulated protein-encoding genes (Table S4). These included 5 transcription factors and 13 ribosomal proteins. The most highly up-regulated gene at all time points, with the largest positive log2 Fold Change of all genes (not just the shared set), encoded a protein containing a DUF3542 domain (DVG80_03545). An adjacent gene also of unknown function (DVG80_ 03546) was the second most universally up-regulated (Figure S7). DUF3542 is an immunoglobulin-like domain that is related to extracellular immunoprotective functions [57], and InterProScan results were consistent with this, indicating that the DUF3542 domain of DVG80_03545 is flanked by a putative signal peptide, and is predicted to lie outside of the cytoplasmic space. Four of six resuscitation-promoting factor (rpf) genes were up-regulated and DE throughout the course of infection (RIA-643 genes 01821, 01260, 05851, 06126). These genes encode peptidoglycan glycosidases and have redundant and overlap** functionality.
Among the 116 universally down-regulated genes, there were 16 transcriptional regulators (Table S4). Five genes involved in both the TCA and glycoxylate cycles, as well as a sixth gene, malate synthase, which is unique to the glycoxylate cycle, were significantly suppressed at all time points during infection, while three additional related genes were DE at only a subset of time points (Table S5). Other TCA cycle genes which were not DE were generally down-regulated (i.e. negative log2FC as compared to baseline) at all time points (Table S5). Three moeY genes involved in molybdopterin biosynthesis (DVG80_01963, 06751, 06752) were part of the core set, while a fourth related gene, a FAD-binding molybdopterin dehydrogenase (DVG80_02060) was significant at 5, 30, and 60 min, and borderline significant (log2FC: -0.94, p-value: 0.06) at 120 min. A universal stress response gene (DVG80_06017) also showed decreased expression at all time points.
WC1 infection modulated expression of genes involved in metal ion homeostasis
Five genes related to membrane transport of metal ions were universally up-regulated as part of the host core set (Table S4; Table S6). This included two of three contiguous genes encoding the substrate-binding protein (DVG80_04450) and permease (DVG80_04452) of a metal ABC transporter. The putative third component, the ATPase (DVG80_04451), was significantly up-regulated at all time points except 5 min, where it was borderline significant (log2FC: 0.87, p-value: 0.05). Based on homology to to ZnuABC of Rhodococcus erythropolis VKPM Ac-1659 [6], and MntABC of Rhodococcus erythropolis PR4 this transporter was predicted to mediate the influx of zinc and/or manganese. A divalent metal cation transporter (DVG80_05365), and an iron transporter homologous to EfeU, and thus predicted to mediate iron influx [58], were also up-regulated. A third related over-expressed gene, DVG80_03824, was homologous to fhuD of Rhodococcus erythropolis VKPM Ac-1659, which encodes the siderophore binding domain of an ABC transporter [6]. Other genes encoding components of siderophore ABC transporters were DE at all timepoints except 5 min; three of these were up-regulated while a fourth was down-regulated (Table S6).
In contrast to core up-regulated genes largely related to iron and metal import, genes involved in metal export and sequestration were strongly down-regulated throughout the time course of infection (Figure S9; Table S6). A gene (DVG80_04092) encoding a UPF0016 domain containing protein had the largest negative log2 fold change at all time points except 120 min, where it was the second largest. UPF0016 domain containing proteins have been shown to be involved in manganese export [59]. The expression of a VIT family protein (DVG80_05717) decreased over the time course of infection, from a log2FC of -2.55 to -4.18 by 120 min (Figure S9; Tables S4 and S6). VIT family proteins are ferritins that export iron and/or manganese, and have been characterized in numerous organisms, including various Rhodococcus species [60], other bacterial lineages [61], fungi [62], eukaryotic parasites [63, 64], and plants [65]. A bacterioferritin (DVG80_04076) was significantly under-expressed at all time points, reaching strongest suppression at 60 min post infection (Figure S4; Table S6). A second bacterioferritin in the RIA-643 genome was not DE, but rather exhibited constitutively low expression levels including at baseline. Similarly, genes homologous to components of an iron-regulated transcriptional repressors, furA (DVG80_03017), furB (DVG80_00409), and ideR (DVG80_01378) were not among down-regulated DE genes. The fur homologs exhibited low transcriptional levels at all time points, while expression of the ideR homolog slightly suppressed at all time points. Cytochrome d ubiquinol oxidase subunit I (DVG_05658) showed a markedly strong drop in expression from 5 min (log2FC: -1.21) to 30 min (log2FC: -4.45), and this decrease continued over time. The adjacent gene encoding subunit II (DVG_05659) was significantly and strongly down-regulated at all time points except 5 min. The cytochrome d oxidase contains iron conjugated structurally in heme [66].
Functional enrichment of host genes reflected the phage WC1 lifecycle
Enriched functional categories changed over the time course, reflecting a transcriptional response to the phases of phage infection (Figs. 5 and 6; Tables S7, S8, S9). At 5 min post-infection, phage WC1 was early in its latent period, beginning replication (Fig. 1A). Enriched terms, pathways, and genes in the host largely corresponded to host stress responses as well as DNA replication (Figs. 5 and 6; Tables S7, S8, S9). The enriched KEGG homologous recombination pathway included homologs of the SOS-response protein RecA (DVG_001424) and RecC (DVG80_03423) of the RecBCD pathway for double-stranded breaks [67]. RecX (DVG80_01423), the transcriptional mediator of RecA, was significantly up-regulated at 5 min as compared to baseline (log2FC: 1.69, p-value: 4.63E-7) as well. Other DNA repair mechanisms associated with the SOS-response were also enriched in both KEGG and GO, including activation of nucleotide excision repair, base excision repair and mismatch repair (Figs. 5 and 6) [67]. While KEGG pathways and GO terms also indicate significant up-regulation of DNA replication, which corresponds to phage takeover of host replication resources, there were also changes in host metabolism. These included significant up-regulation of ribosomal proteins necessary for mRNA translation and increases in histidine catabolism and glutamate biosynthesis (Figs. 5 and 6). Finally, DNA restriction-modification (RM) systems were enriched, which specifically corresponded to five up-regulated components of a Type I RM system (Fig. 6; Table S7).

Enriched KEGG pathways during time course of infection as compared to baseline. The direction of the arrows indicate if the DE genes were up-regulated or down-regulated, and the numeric values indicate the number of genes. Only DE genes with absolute log2 fold change greater than or less than − 0.58 were included in the analysis to reflect a fold-change of 1.5

Enriched GO categories during time course of infection as compared to baseline by ontology. All categories indicated were up-regulated. The numbers inside the circles indicate the median gene count for each category. Categories were obtained as described in methods; the full list of terms in each category and individual gene counts for each term are provided in Supplementary Table S6. Only DE genes with absolute log2 fold change greater than or less than − 0.58 were included in the analysis to reflect a fold-change of 1.5
At 30 min into the infection time course, the host response shifted significantly to increased transcriptional and translational activity in contrast to significant down-regulation of host metabolic pathways (Figs. 5 and 6). This corresponded to the substantial phage replication and protein synthesis occurring during the latent period of WC1 (Fig. 1A). Two KEGG pathways were up-regulated: RNA polymerase and ribosome, the latter of which consisted of 43 ribosomal proteins (Fig. 5). Enriched GO terms also indicate up-regulation of genes involved in gene expression, tRNAs, and ribosomal proteins as well as the formation of the ribosomal complex, translational initiation, and peptide biosynthesis (Fig. 6). Genes associated with the GO category macromolecule biosynthetic process at 30 min were largely ribosomal proteins, sigma factors, and translation elongation factors, but also included the LexA repressor, which is a transcriptional inhibitor of SOS-response related proteins [67]. A sequential comparison between 30 and 5 min (as opposed to baseline) showed significant down-regulation at 30 min of homologous recombination, mismatch repair, and nucleotide excision repair as well as many functions involved in DNA replication that had been enriched at the earlier time point (Figure S10; Table S10). Several KEGG pathways were down-regulated at 30 min as compared to the baseline, including those involved in carbon and energy metabolism, lipid metabolism, and branched-chain amino acid metabolism (Fig. 5). The down-regulated synthesis of secondary metabolites pathway included eight genes involved in the TCA cycle, five of which were part of the core down-regulated set (Table S4; Table S5).
After the end of the WC1 latent period, host transcriptional responses were highly similar to what was observed at 30 min. Direct comparison between 60 and 30 min showed no difference in functional enrichment, i.e., there were no significant KEGG pathways or GO terms when comparing 60 to 30 min (Fig. 5). When compared to baseline, up-regulated functional categories at 60 min were a subset of those at 30 min, predominantly related to transcription and translation (Figs. 5 and 6; Tables S7, S8, S9). Proteasomes were functionally enriched, with both the alpha and beta subunits, which are sufficient to construct the proteasome [68], as well as an associated ATPase up-regulated at 60 min. By 120 min post-infection, many of the functionally enriched host responses closely mirrored those observed at earlier stages of infection. The increase in transcriptional and translational activity from the baseline continued. However, DNA repair mechanisms including components of the SOS response were up-regulated as was endonuclease activity (Fig. 6; Table S7), similar to what was observed at 5 min, which corresponds to a potential second wave of phage infection (Fig. 1A). Oxidative phosphorylation was significantly suppressed at 120 min as compared to baseline, which was not observed at any other time point (Fig. 5).
Discussion
In this study, we characterized WC-1, a new phage of Rhodococcus erythropolis RIA-643. While several Rhodococcus phage have previously been sequenced and annotated [20, 69], global patterns of gene expression during host infection have not yet been sufficiently explored. To the best of our knowledge, our study represents the first exploration of global gene expression following phage infection of a Rhodococcus host. We have described temporal dynamics of both phage and host transcriptomes, which may serve as a model not only for phage-host interactions in R. erythropolis RIA-643, but more generally for other strains and species in and beyond the genus.
WC1 followed an often reported pattern of phage transcriptional activation, expressing temporal classes of early, middle, and late genes [70,71,72,73,74,75,76,77]. These genes also clustered spatially within the genome, with early genes on the right arm, middle genes located more centrally, and late genes on the left arm. Central and left arm genes which could be annotated had regulatory functions and roles in DNA replication, while right arm genes included virion components and accessory factors for assembly. This genomic arrangement is syntenous with other cluster CA phage [20]. Rhodococcal phage have been shown to be polyvalent, with host ranges extending to other genera in some cases [15, 17, 78]. Highly similar phage of Mycobacterium smegmatis [22] as well as divergent phage of Pseudomonas aeruginosa [73] showed preservation of temporal activation patterns across synteny blocks. Taken together, these suggest that other cluster CA phage may follow patterns of expression similar to WC1, and may do so in multiple hosts. Furthermore, M. smegmatis phage displayed similar spatial arrangements and temporal expression patterns to WC1 including highly transcribed RNAs localized to non-coding regions [22, 23]. Three non-coding RNAs located at the far left, middle, and far right of the WC1 were among the most highly expressed transcripts at all time points except 5 min after infection. Similar ncRNAs at both the right and left extremes have been observed in Mycobacteriophages D29 and Kampy, while other cluster A Mycobacteriophage including L5, StarStuff, Redrock and SWU1 contain the right-hand ncRNA only [22, 23]. While the exact function of these noncoding regions is unknown, the region on the far right has been shown to be toxic to growth for both M. smegmatis and E. coli, and potentially essential for lytic growth [22].
Two main strategies for phage takeover of host cells have been previously described. The first involves widespread suppression, where the majority of host genes are down-regulated and host metabolism is shut down early in infection [71, 72, 79,80,81,82]. Alternatively, phage may exert more selective control, preserving the host’s regulatory mechanisms and upregulating more genes than inhibiting to create an environment most conducive for virion production [98]. During WC1 infection, we saw increased expression of metal ion ABC transporters including those mediated by siderophores as well as decreased expression of metal ion exporters and bacterioferritin. This was coupled with low expression throughout infection of a homolog to the iron-binding furAB repressor, and non-DE suppression of expression of a putative iron-binding ideR repressor. First described in E. coli, fur family repressors bind iron in times of sufficient supply to suppress transcription of genes related to iron import [99]. The ideR repressor also binds iron and regulates responses to oxidative stress in M. tuberculosis, and has also been shown to be present in other strains of R. erythropolis as well as active in R. equi [100]. The combination of increased import and decreased export and sequestration with a lack of transcriptional repression which would be indicative of high iron conditions suggests selective phage-mediated control to increase intracellular iron concentrations to facilitate virion replication and assembly. We also note the presence of a phage gene encoding a Cas exonuclease, however no identifiable CRISPR systems are present in the host genome, a result reported for other Rhodococcus species [101, 102].
There are apparent limitations of the current study, including the large number of phage and host gene products of unknown function. As genomic and metagenomic sequencing efforts continue en masse, future characterization of these proteins may become possible. Phage WC1 bears strong identity to known temperate phage, and contains integrase and excise, hence indicating that it is capable of lysogeny. We observed that these genes had constitutively low expression during the infection time course. In order to search for alignments between the phage and host genomes, the phage genome was processed into non-overlap** 1000 bp pieces. Analysis with blastn showed no alignments meeting default non-stringent BLAST thresholds (E-value cutoff of 10). However, in the current study, we do not know what proportion of host cells may be lysogens, and further to that, how many may be uninfected. Finally, further exploration with downstream ‘omics, namely proteomics and metabolomics, will be necessary to confirm these results as well as shed further light on interactions beyond the transcriptional level.
Here, we have shed light on previously undescribed transcriptional dynamics during phage infection of a Rhodococcus host, using the novel phage WC1. Given the high level of similarity between phage of cluster CA, and polyvalence of Rhodococcus phage, results from this system may have broader applicability to predator-prey dynamics in a range of environmentally, industrially, and biomedically important bacterial hosts. Further elucidation of the interplay between Rhodococcus phage and their hosts will be essential for the advancement of phage-mediated biocontrol strategies.
Conclusions
The Rhodococcus genus is well recognized for its ability to synthesize valuable compounds, particularly steroids, as well as its capacity to degrade a wide range of harmful environmental pollutants. A detailed understanding of these phage-host interactions and gene expression is not only essential for understanding the ecology of this important genus, but will also facilitate development of phage-mediated strategies for bioremediation as well as biocontrol in industrial processes and biomedical applications. Given the current lack of detailed global gene expression studies on any Rhodococcus species, our study addresses a pressing need to identify tools and genes, such as F6 and rpf, that can enhance the capacity of Rhodococcus species for bioremediation, biosynthesis and pathogen control. While host gene expression declined over the course of infection, our results indicate that phage may exert more selective control, preserving the host’s regulatory mechanisms to create an environment conducive for virion production.
Data availability
The complete genome sequence of WC1 was deposited in Genbank and is available via accession number MZ402608. RNA-Sequencing data can be accessed via NCBI’s Short Read Archive at project accession number SRP154435.
Abbreviations
- SEA-PHAGES:
-
Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science
- WC1:
-
Winter Compost 1
- DE:
-
Differential expression/Differentially Expressed
References
Bell K, Philp J, Aw D, Christofi N. A review - the genus Rhodococcus. J Appl Microbiol. 1998;85:195–210.
Ivshina I, Bazhutin G, Tyumina E. Rhodococcus strains as a good biotool for neutralizing pharmaceutical pollutants and obtaining therapeutically valuable products: through the past into the future. Front Microbiol. 2022;13:967127.
Kim D, Choi KY, Yoo M, Zylstra GJ, Kim E. Biotechnol Potential Rhodococcus Biodegradative Pathways. 2018;28:1037–51.
Guevara G, Flores YO, Heras LF, de las, Perera J, Llorens JMN. Metabolic engineering of Rhodococcus ruber Chol-4: a cell factory for testosterone production. PLoS ONE. 2019;14:e0220492.
Zappaterra F, Costa S, Summa D, Bertolasi V, Semeraro B, Pedrini P, Buzzi R, Vertuani S. Biotransformation of Cortisone with Rhodococcus rhodnii: synthesis of new steroids. Molecules. 2021;26:1352.
Korzhenkov AA, Bakhmutova ED, Izotova AO, Bavtushnyi AA, Sidoruk KV, Patrusheva EV, Patrushev MV, Toshchakov SV. Draft genome sequence of Rhodococcus erythropolis VKPM Ac-1659, a putative oil-degrading strain isolated from Polluted Soil in Siberia. Microbiol Resour Announc 10:e00535–21.
Ehsani E, Jauregui R, Geffers R, Jareck M, Boon N, Pieper DH, Vilchez-Vargas R. Draft genome sequence of Rhodococcus sp. Strain 311R. Genome Announc. 2015;3. https://doi.org/10.1128/genomea.00378-15.
Ohhata N, Yoshida N, Egami H, Katsuragi T, Tani Y, Takagi H. An extremely oligotrophic bacterium, Rhodococcus erythropolis N9T-4, isolated from Crude Oil. J Bacteriol. 2007;189:6824–31.
Cornelis K, Ritsema T, Nijsse J, Holsters M, Goethals K, Jaziri M. The plant pathogen Rhodococcus fascians colonizes the exterior and interior of the aerial parts of plants. Mol Plant-Microbe Interact MPMI. 2001;14:599–608.
Vázquez-Boland JA, Meijer WG. The pathogenic actinobacterium Rhodococcus equi: what’s in a name? Mol Microbiol. 2019;112:1–15.
Baba H, Nada T, Ohkusu K, Ezaki T, Hasegawa Y, Paterson DL. First case of bloodstream infection caused by Rhodococcus erythropolis. J Clin Microbiol. 2009;47:2667–9.
Stewart A, Sowden D, Caffery M, Bint M, Broom J. Rhodococcus equi infection: a diverse spectrum of disease. IDCases. 2019;15:e00487.
Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–84.
Gill JJ, Wang B, Sestak E, Young R, Chu K-H. Characterization of a Novel Tectivirus Phage Toil and its potential as an Agent for Biolipid extraction. Sci Rep. 2018;8:1062.
Petrovski S, Seviour RJ, Tillett D. Characterization of the genome of the polyvalent lytic bacteriophage GTE2, which has potential for biocontrol of Gordonia-, Rhodococcus-, and Nocardia-stabilized foams in activated sludge plants. Appl Environ Microbiol. 2011;77:3923–9.
Petrovski S, Batinovic S, Rose Jja, Seviour Rj. Biological control of problematic bacterial populations causing foaming in activated sludge wastewater treatment plants—phage therapy and beyond. Lett Appl Microbiol. 2022;75:776–84.
Shibayama Y, Dabbs ER. Phage as a source of antibacterial genes. Bacteriophage. 2011;1:195–7.
Russell DA, Hatfull GF. PhagesDB: the actinobacteriophage database. Bioinforma Oxf Engl. 2017;33:784–6.
Hanauer DI, Graham MJ, Betancur SEA-PHAGES, Bobrownicki L, Cresawn A, Garlena SG, Jacobs-Sera RA, Kaufmann D, Pope N, Russell WH, Jacobs DA, Sivanathan WR, Asai V, Hatfull DJ GF. An inclusive Research Education Community (iREC): impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci. 2017;114:13531–6.
Bonilla JA, Isern S, Findley AM, Klyczek KK, Michael SF, Saha MS, Buchser WJ, Forsyth MH, Paudel S, Gissendanner CR, Wiedemeier AMD, Alonzo FL, Garlena RA, Russell DA, Pope WH, Cresawn SG, Jacobs-Sera D, Hatfull GF. Genome sequences of 19 Rhodococcus erythropolis Cluster CA Phages. Genome Announc. 2017;5:e01201–17.
Ponce Reyes S, Park PJ, Kaluka D, Washington JM. Complete genome sequence of Rhodococcus erythropolis Phage Shuman. Microbiol Resour Announc. 2019;8:e00113–19.
Dedrick RM, Mavrich TN, Ng WL, Hatfull GF. Expression and evolutionary patterns of mycobacteriophage D29 and its temperate close relatives. BMC Microbiol. 2017;17:225.
Halleran A, Clamons S, Saha M. Transcriptomic characterization of an infection of Mycobacterium smegmatis by the cluster A4 Mycobacteriophage Kampy. PLoS ONE. 2015;10:e0141100.
Fan X, Duan X, Tong Y, Huang Q, Zhou M, Wang H, Zeng L, Young RF, **e J. The global reciprocal reprogramming between Mycobacteriophage SWU1 and Mycobacterium reveals the Molecular Strategy of Subversion and Promotion of phage infection. Front Microbiol. 2016;7:41.
Simon Andrews. 2010. FastQC: a quality control tool for high throughput sequence data.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202.
The Actinobacteriophage Database. | Protocols for Sequencing. https://phagesdb.org/workflow/Sequencing/. Retrieved 19 July 2023.
JG Lawrence. 2007. DNA Master.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinforma Oxf Engl. 2007;23:673–9.
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–18.
Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–6.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol Clifton NJ. 2019;1962:1–14.
Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics. 2011;12:395.
Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–31.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genoty** with HISAT2 and HISAT-genotype. 8. Nat Biotechnol. 2019;37:907–15.
Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. BamTools: a C + + API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–2.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–94.
Schurch NJ, Schofield P, Gierliński M, Cole C, Sherstnev A, Singh V, Wrobel N, Gharbi K, Simpson GG, Owen-Hughes T, Blaxter M, Barton GJ. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA. 2016;22:839–51.
eggNOG-mapper v2. Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale | Molecular Biology and Evolution | Oxford Academic. https://academic.oup.com/mbe/article/38/12/5825/6379734. Retrieved 21 July 2023.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–20.
Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, Bileschi ML, Bork P, Bridge A, Colwell L, Gough J, Haft DH, Letunić I, Marchler-Bauer A, Mi H, Natale DA, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A. InterPro in 2022. Nucleic Acids Res. 2023;51:D418–27.
Pertea G, Pertea M. 2020. GFF utilities: GffRead and GffCompare. F1000Research 9:ISCB Comm J-304.
Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot. Methods Mol Biol Clifton NJ. 2007;406:89–112.
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–41.
Frontiers | Summary Visualizations of Gene Ontology Terms With GO-Figure!. https://www.frontiersin.org/articles/https://doi.org/10.3389/fbinf.2021.638255/full. Retrieved 9 August 2023.
Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, Alcoser TA, Alexander LM, Alfano MB, Alford ST, Amy NE, Anderson MD, Anderson AG, Ang AAS, Ares M, Barber AJ, Barker LP, Barrett JM, Barshop WD, Bauerle CM, Bayles IM, Belfield KL, Best AA, Borjon A, Bowman CA, Boyer CA, Bradley KW, Bradley VA, Broadway LN, Budwal K, Busby KN, Campbell IW, Campbell AM, Carey A, Caruso SM, Chew RD, Cockburn CL, Cohen LB, Corajod JM, Cresawn SG, Davis KR, Deng L, Denver DR, Dixon BR, Ekram S, Elgin SCR, Engelsen AE, English BEV, Erb ML, Estrada C, Filliger LZ, Findley AM, Forbes L, Forsyth MH, Fox TM, Fritz MJ, Garcia R, George ZD, Georges AE, Gissendanner CR, Goff S, Goldstein R, Gordon KC, Green RD, Guerra SL, Guiney-Olsen KR, Guiza BG, Haghighat L, Hagopian GV, Harmon CJ, Harmson JS, Hartzog GA, Harvey SE, He S, He KJ, Healy KE, Higinbotham ER, Hildebrandt EN, Ho JH, Hogan GM, Hohenstein VG, Holz NA, Huang VJ, Hufford EL, Hynes PM, Jackson AS, Jansen EC, Jarvik J, Jasinto PG, Jordan TC, Kasza T, Katelyn MA, Kelsey JS, Kerrigan LA, Khaw D, Kim J, Knutter JZ, Ko C-C, Larkin GV, Laroche JR, Latif A, Leuba KD, Leuba SI, Lewis LO, Loesser-Casey KE, Long CA, Lopez AJ, Lowery N, Lu TQ, Mac V, Masters IR, McCloud JJ, McDonough MJ, Medenbach AJ, Menon A, Miller R, Morgan BK, Ng PC, Nguyen E, Nguyen KT, Nguyen ET, Nicholson KM, Parnell LA, Peirce CE, Perz AM, Peterson LJ, Pferdehirt RE, Philip SV, Pogliano K, Pogliano J, Polley T, Puopolo EJ, Rabinowitz HS, Resiss MJ, Rhyan CN, Robinson YM, Rodriguez LL, Rose AC, Rubin JD, Ruby JA, Saha MS, Sandoz JW, Savitskaya J, Schipper DJ, Schnitzler CE, Schott AR, Segal JB, Shaffer CD, Sheldon KE, Shepard EM, Shepardson JW, Shroff MK, Simmons JM, Simms EF, Simpson BM, Sinclair KM, Sjoholm RL, Slette IJ, Spaulding BC, Straub CL, Stukey J, Sughrue T, Tang T-Y, Tatyana LM, Taylor SB, Taylor BJ, Temple LM, Thompson JV, Tokarz MP, Trapani SE, Troum AP, Tsay J, Tubbs AT, Walton JM, Wang DH, Wang H, Warner JR, Weisser EG, Wendler SC, Weston-Hafer KA, Whelan HM, Williamson KE, Willis AN, Wirtshafter HS, Wong TW, Wu P, Yang Y jeong, Yee BC, Zaidins DA, Zhang B, Zúniga MY, Hendrix RW, Hatfull GF. Expanding the Diversity of Mycobacteriophages: Insights into Genome Architecture and Evolution. PLoS ONE. 2011;6:e16329.
Arnvig KB, Young DB. Identification of small RNAs in Mycobacterium tuberculosis. Mol Microbiol. 2009;73:397–408.
Grigorov A, Bychenko O, Salina EG, Skvortsova Y, Mazurova A, Skvortsov T, Kaprelyants A, Azhikina T. Small RNA F6 provides Mycobacterium smegmatis entry into Dormancy. Int J Mol Sci. 2021;22:11536.
Houghton J, Rodgers A, Rose G, D’Halluin A, Kipkorir T, Barker D, Waddell SJ, Arnvig KB. The Mycobacterium tuberculosis sRNA F6 modifies expression of essential chaperonins, GroEL2 and GroES. Microbiol Spectr. 2021;9:e0109521.
Nelson JW, Sudarsan N, Furukawa K, Weinberg Z, Wang JX, Breaker RR. Riboswitches in eubacteria sense the second messenger c-di-AMP. Nat Chem Biol. 2013;9:834–9.
Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci U S A. 2004;101:6421–6.
European Bioinformatics Institute. Domain of unknown function DUF3542. InterPro. https://www.ebi.ac.uk/interpro/entry/InterPro/IPR029051/.
Cao J, Woodhall MR, Alvarez J, Cartron ML, Andrews SC. EfeUOB (YcdNOB) is a tripartite, acid-induced and CpxAR-regulated, low-pH Fe2 + transporter that is cryptic in Escherichia coli K-12 but functional in E. Coli O157:H7. Mol Microbiol. 2007;65:857–75.
Zeinert R, Martinez E, Schmitz J, Senn K, Usman B, Anantharaman V, Aravind L, Waters LS. Structure–function analysis of manganese exporter proteins across bacteria. J Biol Chem. 2018;293:5715–30.
Zappa S, Bauer CE. Iron homeostasis in the Rhodobacter genus. Adv Bot Res. 2013;66. https://doi.org/10.1016/B978-0-12-397923-0.00010-2.
Bradley JM, Svistunenko DA, Wilson MT, Hemmings AM, Moore GR, Brun NEL. Bacterial iron detoxification at the molecular level. J Biol Chem. 2020;295:17602–23.
Sorribes-Dauden R, Peris D, Martínez-Pastor MT, Puig S. Structure and function of the vacuolar Ccc1/VIT1 family of iron transporters and its regulation in fungi. Comput Struct Biotechnol J. 2020;18:3712–22.
Aghabi D, Sloan M, Gill G, Hartmann E, Antipova O, Dou Z, Guerra AJ, Carruthers VB, Harding CR. The vacuolar iron transporter mediates iron detoxification in Toxoplasma Gondii. 1. Nat Commun. 2023;14:3659.
A vacuolar iron. -transporter homologue acts as a detoxifier in Plasmodium | Nature Communications. https://www.nature.com/articles/ncomms10403. Retrieved 5 August 2023.
Gollhofer J, Timofeev R, Lan P, Schmidt W, Buckhout TJ. Vacuolar-Iron-Transporter1-Like proteins Mediate Iron Homeostasis in Arabidopsis. PLoS ONE. 2014;9:e110468.
Borisov VB, Siletsky SA, Paiardini A, Hoogewijs D, Forte E, Giuffrè A, Poole RK. Bacterial oxidases of the cytochrome bd family: Redox Enzymes of Unique Structure, function, and Utility as Drug targets. Antioxid Redox Signal. 2021;34:1280–318.
Maslowska KH, Makiela-Dzbenska K, Fijalkowska IJ. The SOS system: a complex and tightly regulated response to DNA damage. Environ Mol Mutagen. 2019;60:368–84.
Subunit topology of the Rhodococcus. proteasome - Zühl – 1997 - FEBS Letters - Wiley Online Library. https://febs.onlinelibrary.wiley.com/doi/full/10.1016/S0014-5793%2896%2901403-2?sid=nlm%3Apubmed. Retrieved 10 August 2023.
Hatfull GF. Actinobacteriophages: Genomics, Dynamics, and applications. Annu Rev Virol. 2020;7:37–61.
Majtan T, Halgasova N, Bukovska G, Timko J. Transcriptional profiling of bacteriophage BFK20: coexpression interrogated by guilt-by-association algorithm. Virology. 2007;359:55–65.
Zhao X, Chen C, Shen W, Huang G, Le S, Lu S, Li M, Zhao Y, Wang J, Rao X, Li G, Shen M, Guo K, Yang Y, Tan Y, Hu F. Global transcriptomic analysis of interactions between Pseudomonas aeruginosa and Bacteriophage PaP3. 1. Sci Rep. 2016;6:19237.
Li T, Zhang Y, Dong K, Kuo C-J, Li C, Zhu Y-Q, Qin J, Li Q-T, Chang Y-F, Guo X, Zhu Y. Isolation and characterization of the Novel phage JD032 and global transcriptomic response during JD032 infection of Clostridioides Difficile Ribotype 078. mSystems. 2020;5:e00017–20.
Blasdel BG, Chevallereau A, Monot M, Lavigne R, Debarbieux L. Comparative transcriptomics analyses reveal the conservation of an ancestral infectious strategy in two bacteriophage genera. ISME J. 2017;11:1988–96.
Lavigne R, Lecoutere E, Wagemans J, Cenens W, Aertsen A, Schoofs L, Landuyt B, Paeshuyse J, Scheer M, Schobert M, Ceyssens P-J. A multifaceted study of Pseudomonas aeruginosa Shutdown by Virulent Podovirus LUZ19. mBio. 2013;4. https://doi.org/10.1128/mbio.00061-13.
Morimoto D, Kimura S, Sako Y, Yoshida T. 2018. Transcriptome analysis of a Bloom-Forming Cyanobacterium Microcystis aeruginosa during Ma-LMM01 phage infection. Front Microbiol 9.
Chevallereau A, Blasdel BG, Smet JD, Monot M, Zimmermann M, Kogadeeva M, Sauer U, Jorth P, Whiteley M, Debarbieux L, Lavigne R. Next-generation -omics approaches reveal a massive alteration of host RNA metabolism during bacteriophage infection of Pseudomonas aeruginosa. PLOS Genet. 2016;12:e1006134.
Li X, Zhang C, ** X, Wei F, Yu F, Call DR, Zhao Z. Temporal transcriptional responses of a Vibrio alginolyticus strain to Podoviridae phage HH109 revealed by RNA-Seq. mSystems 7:e00106–22.
Petrovski S, Dyson ZA, Seviour RJ, Tillett D. Small but sufficient: the Rhodococcus Phage RRH1 has the smallest known Siphoviridae Genome at 14.2 Kilobases. J Virol. 2012;86:358–63.
Mojardín L, Salas M. Global transcriptional analysis of virus-host interactions between phage ϕ29 and Bacillus subtilis. J Virol. 2016;90:9293–304.
Leskinen K, Blasdel BG, Lavigne R, Skurnik M. RNA-Sequencing reveals the progression of phage-host interactions between φR1-37 and Yersinia enterocolitica. Viruses. 2016;8:111.
Kutter E, Bryan D, Ray G, Brewster E, Blasdel B, Guttman B. From host to phage metabolism: hot tales of phage T4’s takeover of E. Coli. Viruses. 2018;10:387.
Wu D, Yuan Y, Liu P, Wu Y, Gao M. Cellular responses in Bacillus thuringiensis CS33 during bacteriophage BtCS33 infection. J Proteom. 2014;101:192–204.
Sacher JC, Flint A, Butcher J, Blasdel B, Reynolds HM, Lavigne R, Stintzi A, Szymanski CM. Transcriptomic analysis of the Campylobacter jejuni response to T4-Like Phage NCTC 12673 infection. Viruses. 2018;10:332.
Yang Z, Yin S, Li G, Wang J, Huang G, Jiang B, You B, Gong Y, Zhang C, Luo X, Peng Y, Zhao X. Global transcriptomic analysis of the interactions between phage φAbp1 and extensively drug-resistant Acinetobacter baumannii. mSystems. 2019;4. https://doi.org/10.1128/msystems.00068-19.
Campoy S, Hervàs A, Busquets N, Erill I, Teixidó L, Barbé J. Induction of the SOS response by bacteriophage lytic development in Salmonella enterica. Virology. 2006;351:360–7.
Howard-Varona C, Lindback MM, Bastien GE, Solonenko N, Zayed AA, Jang H, Andreopoulos B, Brewer HM, del Glavina T, Adkins JN, Paul S, Sullivan MB, Duhaime MB. Phage-specific metabolic reprogramming of virocells. 4. ISME J. 2020;14:881–95.
Osterhout RE, Figueroa IA, Keasling JD, Arkin AP. Global analysis of host response to induction of a latent bacteriophage. BMC Microbiol. 2007;7:82.
Mukamolova GV, Murzin AG, Salina EG, Demina GR, Kell DB, Kaprelyants AS, Young M. Muralytic activity of Micrococcus luteus Rpf and its relationship to physiological activity in promoting bacterial growth and resuscitation. Mol Microbiol. 2006;59:84–98.
Downing KJ, Mischenko VV, Shleeva MO, Young DI, Young M, Kaprelyants AS, Apt AS, Mizrahi V. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect Immun. 2005;73:3038–43.
Kana BD, Mizrahi V. Resuscitation-promoting factors as lytic enzymes for bacterial growth and signaling. FEMS Immunol Med Microbiol. 2010;58:39–50.
Wang Y, Shi J, Tang L, Zhang Y, Zhang Y, Wang X, Zhang X. Evaluation of rpf protein of Micrococcus luteus for cultivation of soil actinobacteria. Syst Appl Microbiol. 2021;44:126234.
Ealand C, Rimal B, Chang J, Mashigo L, Chengalroyen M, Mapela L, Beukes G, Machowski E, Kim SJ, Kana B. Resuscitation-promoting factors are required for Mycobacterium smegmatis Biofilm formation. Appl Environ Microbiol. 2018;84:e00687–18.
Meeske AJ, Nakandakari-Higa S, Marraffini LA. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. 7760. Nature. 2019;570:241–5.
Bacterial iron homeostasis | FEMS Microbiology Reviews | Oxford Academic. https://academic.oup.com/femsre/article/27/2-3/215/614497. Retrieved 6 August 2023.
Anjem A, Varghese S, Imlay JA. Manganese import is a key element of the OxyR response to hydrogen peroxide in Escherichia coli. Mol Microbiol. 2009;72:844–58.
Que Q, Helmann JD. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol Microbiol. 2000;35:1454–68.
Bonnain C, Breitbart M, Buck KN. 2016. The Ferrojan Horse hypothesis: Iron-virus interactions in the Ocean. Front Mar Sci 3.
Wicke L, Ponath F, Coppens L, Gerovac M, Lavigne R, Vogel J. Introducing differential RNA-seq map** to track the early infection phase for Pseudomonas phage ɸKZ. RNA Biol. 2021;18:1099–110.
Bagg A, Neilands JB. Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry. 1987;26:5471–7.
Boland CA, Meijer WG. The iron dependent regulatory protein IdeR (DtxR) of Rhodococcus equi. FEMS Microbiol Lett. 2000;191:1–5.
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–51.
Guevara G, Castillo Lopez M, Alonso S, et al. New insights into the genome of Rhodococcus ruber strain Chol-4. BMC Genomics. 2019;20:332.
Acknowledgements
We thank Nick Sheft for related explorations of these data. We also thank everyone in the SEAPHAGES program for their helpful and ongoing assistance.
Funding
This work was supported in part by NIH awards 1 R15 HD077624-01 and 1R15HD114135-01 to MSS.
Author information
Authors and Affiliations
Contributions
DW conducted bioinformatic data analysis, made all the final figures, and played the major role in writing the paper. AH and VG conducted the wet lab experiments. SP analyzed and annotated the WC1 sequence. GS contributed to data analysis. MSS conceptualized and directed the project, oversaw all aspects of the project, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
No applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Willner, D.L., Paudel, S., Halleran, A.D. et al. Transcriptional dynamics during Rhodococcus erythropolis infection with phage WC1. BMC Microbiol 24, 107 (2024). https://doi.org/10.1186/s12866-024-03241-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03241-4