
Abstract
Malaria is a persistent illness that is still a public health issue. On the other hand, marine organisms are considered a rich source of anti‑infective drugs and other medically significant compounds. Herein, we reported the isolation of the actinomycete associated with the Red Sea sponge Callyspongia siphonella. Using "one strain many compounds" (OSMAC) approach, a suitable strain was identified and then sub-cultured in three different media (M1, ISP2 and OLIGO). The extracts were evaluated for their in-vitro antimalarial activity against Plasmodium falciparum strain and subsequently analyzed by Liquid chromatography coupled with high-resolution mass spectrometry (LC-HR-MS). In addition, MetaboAnalyst 5.0 was used to statistically analyze the LC-MS data. Finally, Molecular docking was carried out for the dereplicated metabolites against lysyl-tRNA synthetase (PfKRS1). The phylogenetic study of the 16S rRNA sequence of the actinomycete isolate revealed its affiliation to Streptomyces genus. Antimalarial screening revealed that ISP2 media is the most active against Plasmodium falciparum strain. Based on LC-HR-MS based metabolomics and multivariate analyses, the static cultures of the media, ISP2 (ISP2-S) and M1 (M1-S), are the optimal media for metabolites production. OPLS-DA suggested that quinone derivatives are abundant in the extracts with the highest antimalarial activity. Fifteen compounds were identified where eight of these metabolites were correlated to the observed antimalarial activity of the active extracts. According to molecular docking experiments, saframycin Y3 and juglomycin E showed the greatest binding energy scores (-6.2 and -5.13) to lysyl-tRNA synthetase (PfKRS1), respectively. Using metabolomics and molecular docking investigation, the quinones, saframycin Y3 (5) and juglomycin E (1) were identified as promising antimalarial therapeutic candidates. Our approach can be used as a first evaluation stage in natural product drug development, facilitating the separation of chosen metabolites, particularly biologically active ones.
Similar content being viewed by others
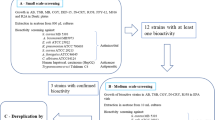


Introduction
Malaria is a long-standing disease and remains a public health problem that most likely originating in Africa [1]. It affects 30 million people globally and kills about 2.5 million people each year, mostly children under the age of five. Chloroquine-resistant Plasmodium falciparum strains are now ubiquitous in all endemic locations around the world [2], and there is also evidence of the emergence of resistance against artemisinin-based combination therapies, which now serve as first line drugs for the treatment of P. falciparum [3]. Therefore, there is a growing need to identify new drugs. Marine ecosystems are composed of taxonomically and biologically diverse macro- and microorganisms that can withstand pressure, salinity, and temperature extremes. They are capable of producing unique compounds with novel therapeutic applications that aren't found in their terrestrial equivalents [4]. Sponges (phylum Porifera) are among the oldest and most abundant multicellular marine organisms. The Red Sea sponge, Callyspongia siphonella (family Callyspongiidae) is found in shallow water throughout the Gulfs of Aqaba and Suez mostly deeper than 5 m [5]. On the other hand, microbial populations, including archaea, bacteria, fungus and viruses abound in the interiors of different sponge species [6]. So far, at least 32 bacterial phyla and candidate groups have been identified in marine sponges, including Acidobacteria, Actinobacteria, Chloroflexi, Nitrospira, Cyanobacteria, Bacteriodetes, Gemmatimonadetes, Planctomycetes, Spirochaetes and Proteobacteria. Actinomycetes, particularly those associated with marine sponges are well-known for their capacity to generate novel lead molecules including phenazines, peptides and alkaloids of clinical and pharmacological significance, such as: antibacterial, antifungal, antiparasitic, immunomodulatory, anti-inflammatory, antioxidant, and anticancer activities [6,7,8]. However, many secondary metabolites encoded in actinomycete genomes are yet to be found, owing to the fact that these genes are not transcribed under standard laboratory conditions. The "one strain many compounds" (OSMAC) strategy involves manipulating fermentation conditions and is an efficient way to activate metabolic pathways that are either silent or poorly expressed [9]. In the present study, the actinomycete Streptomyces associated with the Red Sea sponge C. siphonella was isolated, identified and cultured in various culture media utilizing the OSMAC approach to determine the optimal media for the production of beneficial secondary metabolites. Antimalarial screening was performed to assess the effectiveness of Streptomyces species fermented extracts against P. falciparum strain. LC-HR-MS assisted chemical investigation followed by multivariate data statistical analysis (MVDA) was carried out in order to reduce the amount of data collected and find correlation and differentiation among the samples tested. Finally, molecular docking analysis was performed for the suggested compounds against the malarial active site, lysyl-tRNA synthetase (PfKRS1).
Material and methods
Sponge collection
The Red Sea sponge Callyspongia siphonella (1.5 kg) was obtained in March 2021 at a depth of 7 m off the coast of Hurghada on the Red Sea (2,715,048″ north (N), 334,903″ east (E)). The Invertebrates Department, National Institute of Oceanography and Fisheries, Red Sea Branch, Hurghada, Egypt, was given a voucher sample (NIOF730/2021). The sponge was identified by El-Sayd Abed El-Aziz (Department of Invertebrates Lab., National Institute of Oceanography and Fisheries, Red Sea Branch, 84511 Hurghada, Egypt). Sponge biomass was transferred to plastic bag containing seawater and transported to the laboratory. Sponge specimens were rinsed in sterile seawater, cut into pieces of ca. ˜1 cm3, and then thoroughly homogenized in a sterile mortar with 10 volumes of sterile seawater. The supernatant was diluted in ten-fold series (10-1, 10-2, 10-3) and subsequently plated out on agar plates.
Actinomycetes isolation
Three different media [M1, ISP-2 medium and Oligotrophic medium (OLIGO)] were used for isolation of actinobacteria. All media were supplemented with 0.2 µm pore size filtered cycloheximide (100 µg/mL), nystatin (25 µg/mL) and nalidixic acid (25 µg/mL) to facilitate the isolation of slow-growing actinobacteria. Cycloheximide and nystatin inhibit fungal growth, while nalidixic acid inhibits many fast-growing Gram-negative bacteria [8]. All media contained Difco Bacto agar (18 g/L) and were prepared in 1 L artificial sea water (NaCl 234.7 g, MgCl2.6 H2O 106.4 g, Na2SO4 39.2 g, CaCl2 11.0 g, NaHCO3 1.92 g, KCl 6.64 g, KBr 0.96 g, H3BO3 0.26 g, SrCl2 0,24 g, NaF 0.03 g and ddH2O to 10.0 L). The inoculated plates were incubated at 30 °C for 6–8 weeks. Distinct colony morphotypes were picked and re-streaked until visually free of contaminants. 30 isolates were picked up upon morphological differences. The isolates were maintained on plates for short-term storage, and long-term strain collections were set up in medium supplemented with 30% glycerol at −80 °C.
Molecular identification and phylogenetic analysis
16S rRNA gene amplification cloning and sequencing by Real-Time PCR (qRT-PCR) were performed as described in Cheng et al., (2015) [10] using the universal primers 27F and 1492R (Goldstar, Eurogentec, Seraing, Belgium) [11]. Chimeric sequences were identified by using the Pintail program [12]. The genus-level affiliation of the sequence was validated using the Ribosomal Database Project Classifier. The genus-level identification of all the sequences was done with RDP Classifier (-g 16srrna, -f allrank) and validated with the SILVA Incremental Aligner (SINA) (search and classify option). An alignment was calculated again using the SINA web aligner (variability profile: bacteria) [13]. Gap-only position were removed with trimAL (-noallgaps). For phylogenetic tree construction, the best fitting model was estimated initially with Model Generator. RAxML (-f a -m GTRGAMMA –x 12345 –p 12345 -# 1000) and the estimated model was used with 1000 bootstrap resamples to generate the maximum-likelihood tree [14]. Visualization was done with TreeGraph3 [15].
Phylogenetic analysis
Nucleotide sequences of 16S rRNA of type strains of Streptomyces species and the outgroup Nocardioides sediminis were extracted from EZTAXON database https://www.ezbiocloud.net/ . The sequences from Streptomyces type strains were chosen based on the highest percentage of similarity against the query sequence sequenced in this study (Strain US4). The 16S rRNA sequences were aligned using Muscle (10 maximum number of iterations) implemented in Geneious Prime 2022.1.1. https://www.geneious.com/. The alignment was manually curated, insertions were trimmed and the best nucleotide model determined using MEGAX [16]. Maximum Likelihood tree was computed using MEGA X with 1000 bootstrap replicates and using the best model (TN93+G+I = Tamura Nei + Gamma Distribution + Invariant sites). The accession number for the 16S rRNA gene sequence of strain US4 is: OQ739151.
Extract preparation
The isolated strain was cultivated using three different production media (M1, ISP2, OLIGO), static and shaking approaches to produce 6 organic extracts. The liquid cultures were grown for 10 days at 30 °C while shaking at 150 rpm. The culture was then filtered, and supernatant was extracted with ethyl acetate, while the cells and mycelia were extracted by shaking with methanol for 4 hours. The ethyl acetate extracts were stored at 4 °C.
LC-MS profiling
Samples were prepared and analyzed as previously described by Gamaleldin et al. [8]. For mass spectrometry analysis, three extracts from the samples were produced using ethyl acetate and three using methanol, both at a concentration of 1 mg/mL. According to Abdelmohsen et al. 2014 [17], the recovered ethyl acetate extracts were subjected to metabolic investigation using LC-HR-ESI-MS. We employed a Synapt G2 HDMS quadrupole time-of-flight hybrid mass spectrometer (Waters, Milford, USA) coupled to an Acquity Ultra Performance Liquid Chromatography system. On an Accela HPLC (Thermo FisherFisher Scientific, Bremen, Germany), extracts diluted to 1 mg/mL were examined in triplicates. The HPLC column was an ACE (Hichrom Limited, Reading, UK) C18, 75 mm x 3.0 mm, 5 µm column. The mobile phase composed of purified water (A) and acetonitrile (B), each containing 0.1% formic acid. The gradient programme began with 10% B, raised B linearly in 30 min to 100% B at a flow rate of 300 µL/min, remained isocratic for 5 min, and then linearly decreased B to 10% B in 1 min. Before the subsequent injection, the column was re-equilibrated with 10% B for 9 min. Each sample's analysis took 45 minutes in total. The injection volume was 10 µL, and 12 °C was kept in the tray temperature [18]. The high-resolution mass spectrometry was performed using both positive and negative ESI ionization modes in conjunction with a spray voltage of 4.5 kV, a capillary temperature of 320 °C, and a mass range of m/z 150–1500. Based on the predetermined parameters, the MS dataset was analyzed, and data were retrieved using Mzmine 2.20. Chromatogram builder and chromatogram deconvolution were detected alongside mass ion peak detection. The isotopic peaks of grouper were used to differentiate isotopes, and the local minimum search algorithm was addressed. Using the gap-filling peak finder, missing peaks were highlighted. A complicated search and an adduct search were also conducted. Afterwards, peak identification and the prediction of the chemical formula were applied to the processed data set. The corresponding extract's positive and negative ionization mode data sets were dereplicated and compared to the DNP (Dictionary of Natural Products) databases. ChemBioDraw Ultra 14.0 software was used to create chemical structure drawings.
Multivariate and statistical analysis
Data was prepared as illustrated by El-Hawary et al. [19] and uploaded to MetaboAnalyst 5.0 server (https://www.metaboanalyst.ca).
Antimalarial assay
This assay was performed as previously described by Gamaleldin et al. [8]. The Malstat assay, which was described in [12, 13], was used to determine the extract's anti-plasmodial activity on P. falciparum erythrocytic replication in vitro. P. falciparum NF54 strains were used to plate synchronized ring-stage parasites with 1% parasitaemia in triplicate in 96-well plates (200 L/well) in the presence of successive dilutions of extracts diluted in 0.5% v/v dimethyl sulfoxide (DMSO). The extracts were cultured with the parasites for 72 hours at 37 °C with (90 % N2, 5 % O2, and 5 % CO2. The parasites were incubated with DMSO at a concentration of 0.5 % alone as a negative control, and the parasites were incubated with 20% DMSO as a positive control. Afterwards, 20 L was taken out and put on a fresh 96-well microtiter plate with 100 L of the Malstat reagent (1% Triton X-100, 10 mg of l-lactate, 3.3 mg Tris, and 0.33 mg of 3-Acetylpyridine adenine dinucleotide dissolved in 1 mL of distilled water, pH 9.0). A 20 L mixture of NBT (Nitro Blue Tetrazolium)/Diaphorase (1:1; 1 mg/mL stock each) was then added to the Malstat reaction to measure the plasmodial lactate dehydrogenase activity. Using the fifth version of the GraphPad Prism programme, the optical densities were determined at 630 nm, and the IC50 values were computed from variable-slope sigmoidal dose-response curves.
Ligand design and docking
In this study, based on this fact that the ligands structures were not exist in PubChem. Avogadro 1.2.0 [20] was used to design the ligands. Molecular dockings of the selected small molecules were carried out by AutoDock 4.2 [21] in order to investigate the binding affinity of the ligands to the protein. AutoDockTools (ADT) 1.5.6 was utilized to prepare the docking inputs files. All hydrogens were added, and Kolman charges were calculated. Afterward, each non-polar hydrogen was merged with its corresponding carbon atom. Followed by specifying the torsion tree, small molecules were saved in PDBQT format and utilized for local docking. The grid box was adjusted to 60 × 50 × 70 Å points in xyz directions with 375 Å spacing set on the ligand-binding site. The LGA was applied using the default values except for the number of GA runs, which considered 500. Docking was performed on a rigid receptor, and small molecules were regarded as flexible. Later, the binding modes of the complex structures were evaluated by Chimera software [22]. Eventually, the best binding modes were selected.
Results
Phylogenetic analysis of the strain Streptomyces sp. US4
Taxonomic affiliation of the isolated strain (US4) was conducted by comparing 16S rRNA sequences from Streptomyces closest type strains. The sequenced strain US4 has the highest 16S rRNA similarity with the type strain Streptomyces dysideae sharing 96% similarity. A phylogenetic tree including the closest 30 type strains from Streptomyces described species verified the supported clade of these two strains (Fig. 1) with a 100% bootstrap support. The cut-off of 98.63% for 16S rRNA species delineation [23] suggest that the strain US4 could represent a new species within this genus. Further analysis will need to be conducted to describe this strain as a novel species.

Neighbour-joining phylogenetic tree based on 16S rRNA sequences of the actinomycete isolate Streptomyces US4 and related sequences. The accession number of strain Streptomyces sp. US4 is OQ739151
Antimalarial assay
The culture extracts were tested in vitro for their antimalarial activities against the pathogenic, Plasmodium falciparum strain and results were presented in (Table 1). Results revealed that the extracts, ISP2-S and ISP2-L exhibited the highest inhibitory activity with IC50 values 2.7 and 9.8 µg/mL, respectively. While, M1-S and M1-L showed moderate anti-plasmodial activity with IC50 values of 11.5 and 18.4 µg/mL, respectively. However, Oligo-S and Oligo-L were inactive compared to the positive control drug chloroquine (IC50 value: 0.022 µg/mL).
Metabolomics and multivariate data analysis
Unsupervised analysis
The PCA pairwise score graphs and scree plot (Fig. 2) revealed that there are five PCA components (PCs) explained 94.2% of the total variation, in which the first and second PCs individually contributed to 68.7% of the total variation (PC1 and PC2 represent 42.7% and 26%, respectively (Fig. 2B). In PCA 2D scores plot (Fig. 3A), the samples were distributed to three separated areas between PC1 and PC2, which indicated statistically significant differences between the extracts; the outliers were for the samples ISP2-S and M1-S (Fig. 3A). The PCA loadings plot (Fig. 3B) indicated the metabolites (m/z) that contributed to the variation of the anomalous samples. Dictionary of Natural Products (DNP) was used to annotate such metabolites; the annotated discriminatory compounds for ISP2-S corresponding to m/z (retention time in min.) 197.08152 [M_H]_ (2.701041) was identified as; 2-Butyl-4-(hydroxymethyl)-3-furancarboxylic acid (C10H14O4) [24] or; 4-(Hydroxymethyl)-2-(2-methylpropyl)-3-furancarboxylic acid (C10H14O4) [25]. The mass ion peak at m/z 355.1184 [M_H]_ (RT, 3.9897 min), corresponding to the proposed molecular formula C20H20O6, was identified as antibiotic X 14881B [26]. The mass ion peak at m/z 325.10801 [M_H]_ (RT, 4.0799 min), in accordance with the molecular formula C19H18O5 was recognized as 1,3,6-trihydroxy-8-(3-methylbutyl)anthraquinone [27]. Whereas that at m/z 517.1709 [M _ H] + (RT, 4.4354 min) corresponding to the molecular formula C26H28O11 was suggested to be didemethylmutactimycin [28] or 8-demethoxy-2'-de-O-methylsteffimycin D [29], On the other hand, the annotated discriminatory compounds for M1-S are: The mass ion peak corresponding to m/z (retention time in min.) 425.20793 [M_H] + (3.6336) was putatively identified as JBIR 107 [30] or streptophenazine B or streptophenazine A [31]. (C24H28N2O5). The mass ion peak at m/z 439.22284 [M+H] + (RT, 4.2521 min), corresponding to the proposed molecular formula (C25H30N2O5), was identified as streptophenazine F or streptophenazine G [31]. Where that at m/z 375.2021 [M _ H] + (RT, 3.8968 min) corresponding to the molecular formula (C18H30O8) was suggested to be 2-hydroxy, 2,3-dihydro-antibiotic SEN 366D1. Finally, the mass ion peak at 325.2015 [M_H]_ (RT, 3.8514) corresponding to the molecular formula (C18H30O5) was annotated as Albocycline M6 or Albocycline M3 or Antibiotic A121 [32, 33].

A PCA pairwise score plot of the unsupervised method; (B) PCA scree plot of the unsupervised method

Metabolomics multivariate analysis. A 2D PCA scores plot of the unsupervised method; (B) 2D PCA loadings plot of the unsupervised method; (C) HCA plot showed as dendogram; (D) K-means clustering analysis
Clustering analysis
The hierarchical clusters analysis (Fig. 3C) and the K-means clustering analysis (Fig. 3D) also revealed the diversity of the samples, ISP2-S and M1-S. The diagrams showed a unique variation of the samples, ISP2-S and M1-S, which indicating their chemical diversity from other samples and from each other (Fig. 3).
Supervised analysis
Partial Least Squares - Discriminant Analysis (PLS-DA)
The created model performed well in terms of both prediction (predictive power of models, Q2 = 0.9) and performance (model goodness, R2 = 0.99), a high Q2 value is indicative of good predictions (Q2 values were those that were very near to 1.0). The score plot of partial least squares discriminant analysis (PLS‐DA) revealed a noticeable separation between the sample groups indicating a significant change in the metabolite profile (Fig. 4A); the samples ISP2-S and M1-S were outliers. One of the crucial measurements in PLS-DA is the variable Importance in Projection (VIP), which can be seen in Fig. 4B and Table 2. VIP demonstrated the most 15 important features of highest value identified by PLS-DA (Table 2) (Fig. 5).

Metabolomics multivariate analysis. A PLS-DA scores plot; (B) VIP score plot of PLS-DA

Chemical structures of the 15 most important VIPs
The Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA)
OPLS-DA method was carried out to investigate the link between Streptomyces derived extract's anti-plasmodial efficacy and their metabolite profiles. The developed model performed well in terms of performance (R2 = 0.99) and prediction (Q2 = 0.989) (P < 0.5); the best R2 values were those that were extremely near to 1.0 [34]. The most active extracts, with IC50 values less than 10 µg/ml (ISP2-S and ISP2-L) were classified as active anti-plasmodial. Whereas those with higher values (>10 µg/ml) were classified as inactive. The results were presented in OPLS-DA score plot (Fig. 6) (Table 3).

Metabolomics multivariate analysis; (A) OPLS-DA score plot; (B) OPLS-DA S-plot demonstrating the masses of the most important metabolites (1–8) that may be associated with the anti-plasmodial activity
Molecular docking study
Molecular docking study was employed in order to assess the binding modes and calculate the binding affinity between lysyl-tRNA synthetase (PfKRS1) and the selected small molecules. Initially, this protein (PDB code: 1lyl), was docked into the binding site of ten selected ligands respectively including, juglomycin E (1), didemethylmutactimycin (2a), 8-demethoxy-2'-de-O-methylsteffimycin D (2b), 1,3,6-trihydroxy-8-(3-methylbutyl) anthraquinone (3), ω-hydroxyaloesaponarin II (4), saframycin Y3 (5), antibiotic X 14881B (6), 2-butyl-4-(hydroxymethyl)-3-furancarboxylic acid (7a), 4-(hydroxymethyl)-2-(2-methylpropyl)-3-furancarboxylic acid (7b) and nitropyrrolin A (8). The binding energy scores and intermolecular interaction information of 1lyl and selected ligands are tabulated in Table 4. 3D structures of interaction between lysyl-tRNA synthetase and ten selected ligands are shown in Fig. 7.

3D structures of interaction between lysyl-tRNA synthetase and ten selected ligands
All metabolites demonstrated binding affinity to lysyl-tRNA synthetase binding pocket, with energy values ranging from strong (-6.2) to weak (-3.23). Ten compounds were evaluated, and the quinone derivatives saframycin Y3 and juglomycin E exhibited the highest values (-6.2and -5.13, respectively) followed by ω-hydroxyaloesaponarin II with energy score -4.82. While, antibiotic X 14881B ranked the lowest energy score (-3.23) as presented in Table 4.
Discussion
The scientific community is always fascinated by natural products. Natural products now contain distinctive chemical heterogeneity as a result of evolution over millions of years, giving them drug-like qualities and a variety of biological activities. They have offered a number of medicinal lead compounds. A significant number of these lead chemicals were identified from microbial populations. Actinobacteria are a substantial source of medically relevant chemicals, including anti-infective medicines; It contributed over 80% of the antibiotics currently used in medicine, with Streptomyces accounting for 50% of those compounds [35]. As a result of our continuous attempts to find bioactive natural compounds with antimalarial potential, the actinomycete associated with the Red Sea sponge Callyspongia siphonella was isolated and identified as Streptomyces sp. US4 . Using OSMAC approach, three different media (M1, ISP2 and OLIGO) were used to subculture the isolated strain in order to activate silent and poorly expressed gene clusters. Extracts were screened in-vitro against Plasmodium falciparum strain, and since the antimalarial results were effective, in particular for the extract ISP2-S, this encouraged us to explore their chemical constituents using the most popular efficient method, untargeted LC-HR-MS metabolomic profiling. The obtained LC-HR-MS data was statistically treated using MetaboAnalyst- a Multivariate data statistical analysis (MVA) platform- in order to reduce the vast amount of data obtained and detect correlation and differentiation among the tested samples [36, 37]. The unsupervised principal component analysis (PCA) method and the supervised method Partial Least Squares- Discriminant Analysis (PLS-DA) revealed that the culture extracts ISP2-S and M1-S were dispersed from the others indicating there chemical diversity, which is further corroborated by their distinct patterns in the heatmap plot.
The relation between the extracts and their antimalarial activity was investigated using OPLS-DA method. As shown in the OPLS-DA score plot (Fig. 6A) there are clear differences between active and inactive extracts. Where, active extracts clustered together, indicating that they contain related metabolites that could be responsible for their remarkable antiplasmodial activity. The bioactive discriminatory metabolites that may be related to the observed antimalarial activity of the active extracts were predicted using the OPLS-DA-derived S-Plots (Fig. 6B) (Table 3). Eight metabolites were dereplicated using the Dictionary of Natural Products (DNP), with Quinone derivatives (1–5) being the predominant metabolites (Table 3) (Fig. 8). The distribution of these bioactivity-linked metabolites among the extracts studied was depicted by the heatmap in Fig. 9; it was revealed that Juglomycin E (1) (C14H10O7) corresponding to m/z (retention time in min.) 291.04957 [M_H] + (2.1175) is common in the most active extracts, ISP2-S and ISP2-L.

Structures of metabolites (1–5) that were highly correlated to the extracts’ anti-malarial activity

Heat-map indicating the distribution and abundance of quinones (1–5) in the active antimalarial extracts
The aforementioned findings revealed the abundance of quinone derivatives in the most active extracts. Since various studies revealed that drugs containing quinone nuclei exhibited promising anti-malarial activities [1, 28], it was crucial to apply docking analysis to examine these chemical compounds in more detail, determine their mode of action, and look into the connection between the chemical components and the resulting antimalarial activity. On the other hand, the focus on target-based drug discovery and finding targeted antimalarial drugs that treat all stages of the disease became a need. Previously published data revealed the presence of various malarial targets that affects different stages of parasite infection [8], among them, lysyl-tRNA synthetase, [38] which is an enzyme central to protein translation and responsible for protein formation [39]. Earlier studies revealed that lysyl-tRNA synthetase could be considered as an attractive, druggable, antimalarial target that can be selectively inhibits protein synthesis and prevents liver and blood-stage proliferation [34,35,36,37,38]. Therefore, computational prediction – Docking analysis – was performed in order to screen the annotated quinone derivatives against the antimalarial target, lysyl-tRNA synthetase.
Docking analysis is considered an alternative approach to drug discovery and is preferable to conventional drug discovery efforts that is time consuming, and is often implemented first to screen compounds with promising activity against different protein targets and then to identify their specific targets in order to ascertain their mode of action [39, 40]. Interestingly, all selected metabolites showed binding affinities ranging from (-6.2 and -3.23). Saframycin Y3 (5) and juglomycin E (1), two quinone derivatives, showed the highest values (-6.2 and -5.13, respectively), followed by ω-hydroxyaloesaponarin II (4), which had an energy score of -4.82. Therefore, among the variety of chemical components present in the analyzed extracts, the in silico docking method prioritized the putative active metabolites Saframycin Y3 (5) and juglomycin E (1) as promising antimalarial candidates.
Juglomycin E (1) is a derivative of juglomycins [41], which are broad spectrum naphthoquinone antibiotics naturally produced by different Streptomyces species [42, 43]. Saframycin Y3 (5) is a naturally produced active compound belongs to saframycins, which are heterocyclic quinone groups [44]. Saframycins are well known antitumor antibiotics produced by the actinomycetes strain Streptomyces lavendulae [45].
Conclusion
The marine bacterium, Streptomyces sp. was investigated utilizing the established process using LC-HR- MS-based analytical techniques. The OSMAC technique was used to isolate and subculture the actinomycete, Streptomyces associated with the Red Sea sponge Callyspongia siphonella in three distinct media (M1, ISP2, and OLIGO). The extracts were then examined for their antimalarial effectiveness and chemical diversity. The ISP2 extract showed significant antimalarial efficacy. On the other hand, many metabolites of diverse structural types, primarily quinones, have been found as a result of our study of this species. In silico studies revealed that the metabolites saframycin Y3 (5) and juglomycin E (1) have a high and comparable docking score within all other dereplicated molecules. The focus of this work was on the special role of actinomycetes related to marine species as an untapped source of active metabolites for the development of efficient natural antimalarial medicines. Finally, the journey of finding the best drug-like molecules is a difficult and continual process that takes a lot of time, however, this combination of tools and techniques acts as a model for the development of future targeted antimalarial drugs. Therefore, our research focuses on methods for accelerating the development of effective lysyl-tRNA synthetase inhibitors with therapeutic potential, and could be used to guide the creation of novel antimalarial drugs that can inhibit lysyl-tRNA synthetase. We suggest that future isolation and identification efforts to be only focus on the candidates that scored highest energy binding affinities in the docking study.
Availability of data and materials
The whole genome sequences have been submitted to the NCBI/Nucleotide [accession number of strain Streptomyces sp. US4 is OQ739151].
Direct accessible link: https://www.ncbi.nlm.nih.gov/nuccore/OQ739151.1/.
References
Fotie J. Quinones and Malaria. Antiinfect Agents Med Chem. 2006;5:357–66. https://doi.org/10.2174/187152106778520451.
Hussain H, Specht S, Sarite SR, Saeftel M, Hoerauf A, Schulz B, Krohn K. A New Class of Phenazines with Activity against a Chloroquine Resistant Plasmodium Falciparum Strain and Antimicrobial Activity. J Med Chem. 2011;54:4913–7. https://doi.org/10.1021/jm200302d.
Uwimana A, Legrand E, Stokes BH, Ndikumana JLM, Warsame M, Umulisa N, Ngamije D, Munyaneza T, Mazarati JB, Munguti K, et al. Emergence and Clonal Expansion of in Vitro Artemisinin-Resistant Plasmodium Falciparum Kelch13 R561H Mutant Parasites in Rwanda. Nat Med. 2020;26:1602–8. https://doi.org/10.1038/s41591-020-1005-2.
Abdelmohsen UR, Bayer K, Hentschel U. Diversity, Abundance and Natural Products of Marine Sponge-Associated Actinomycetes. Nat Prod Rep. 2014;31:381–99. https://doi.org/10.1039/c3np70111e.
El-Hawary SS, Sayed AM, Mohammed R, Hassan HM, Rateb ME, Amin E, Mohammed TA, El-Mesery M, Muhsinah AB, Alsayari A, et al. Bioactive Brominated Oxindole Alkaloids from the Red Sea Sponge Callyspongia Siphonella. Mar Drugs. 2019;17:1–13. https://doi.org/10.3390/md17080465.
El-Hawary SS, Sayed AM, Mohammed R, Khanfar MA, Rateb ME, Mohammed TA, Hajjar D, Hassan HM, Gulder TAM, Abdelmohsen UR. New Pim-1 Kinase Inhibitor From the Co-Culture of Two Sponge-Associated Actinomycetes. Front Chem. 2018;6:538. https://doi.org/10.3389/fchem.2018.00538.
Hifnawy MS, Hassan HM, Mohammed R, Fouda MM, Sayed AM, Hamed AA, Abouzid SF, Rateb ME, Alhadrami HA, Abdelmohsen UR. Marine Drugs Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora Sp. UR56 and Actinokinespora Sp. EG49. Mar Drugs. 2020;18:243. https://doi.org/10.3390/md18050243.
Gamaleldin NM, Bahr HS, Mostafa YA, Mcallister BF, Zawily A El, Ngwa C J, Pradel G, Hassan H M, Abdelmohsen U R. Metabolomic Profiling , In Vitro Antimalarial Investigation and In Silico Modeling of the Marine Actinobacterium Strain Rhodococcus Sp . UR111 Associated with the Soft Coral. Antibiotics. 2022;11:1–17.
Abdelmohsen UR, Grkovic T, Balasubramanian S, Kamel MS, Quinn RJ, Hentschel U. Elicitation of Secondary Metabolism in Actinomycetes. Biotechnol Adv. 2015;33:798–811. https://doi.org/10.1016/j.biotechadv.2015.06.003.
Cheng C, Macintyre L, Abdelmohsen UR, Horn H, Polymenakou PN, Edrada-Ebel R, Hentschel U. Biodiversity, Anti-Trypanosomal Activity Screening, and Metabolomic Profiling of Actinomycetes Isolated from Mediterranean Sponges. PLoS One. 2015;10:1–21. https://doi.org/10.1371/journal.pone.0138528.
Hentschel U, Schmid M, Wagner M, Fieseler L, Gernert C, Hacker J. Isolation and Phylogenetic Analysis of Bacteria with Antimicrobial Activities from the Mediterranean Sponges Aplysina Aerophoba and Aplysina Cavernicola. FEMS Microbiol Ecol. 2001;35:305–12.
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New Screening Software Shows That Most Recent Large 16S RRNA Gene Clone Libraries Contain Chimeras. Appl Environ Microbiol. 2006;72:5734–41.
Pruesse E, Peplies J, Glöckner FO. SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinformatics. 2012;28:1823–9.
Stamatakis A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics. 2014;30:1312–3.
Letunic I, Bork P. Interactive Tree Of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinformatics. 2007;23:127–8.
Stecher G, Tamura K, Kumar S. Molecular Evolutionary Genetics Analysis (MEGA) for MacOS. Mol Biol Evol. 2020;37:1237–9.
Abdelmohsen UR, Yang C, Horn H, Hajjar D, Ravasi T, Hentschel U. Actinomycetes from Red Sea Sponges: Sources for Chemical and Phylogenetic Diversity. Mar Drugs. 2014;12:2771–89. https://doi.org/10.3390/md12052771.
Abdelmohsen UR, Cheng C, Viegelmann C, Zhang T, Grkovic T, Ahmed S, Quinn RJ, Hentschel U, Edrada-ebel R. Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora Sp. EG49. Mar Drugs. 2014;12:1220–44. https://doi.org/10.3390/md12031220.
El-Hawary SS, Mohammed R, Bahr HS, Attia EZ, El-Katatny MH, Abelyan N, Al-Sanea MM, Moawad AS, Abdelmohsen UR. Soybean-Associated Endophytic Fungi as Potential Source for Anti-COVID-19 Metabolites Supported by Docking Analysis. J Appl Microbiol. 2021;131:1193–211. https://doi.org/10.1111/jam.15031.
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. Adv Math. 2012;4:1–17. https://doi.org/10.1016/j.aim.2014.05.019.
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30:2785–91. https://doi.org/10.1002/jcc.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–12. https://doi.org/10.1002/jcc.20084.
Stackebrandt E. Taxonomic Parameters Revisited: Tarnished Gold Standards. Microbiol Today. 2006;33:152–5.
Wang J, Zhang H, Yang X, Zhou Y, Wang H, Bai H. HS071, a New Furan-Type Cytotoxic Metabolite from Streptomyces Sp. HS-HY-071. J Antibiot (Tokyo). 2008;61:623–6. https://doi.org/10.1038/ja.2008.82.
Corre C, Song L, O’Rourke S, Chater KF, Challis GL. 2-Alkyl-4-Hydroxymethylfuran-3-Carboxylic Acids, Antibiotic Production Inducers Discovered by Streptomyces Coelicolor Genome Mining. Proc Natl Acad Sci U S A. 2008;105:17510–5. https://doi.org/10.1073/pnas.0805530105.
Parker KA, Ding QJ. A General Approach to Angucyclines: Synthesis of Hatomarubigin A, Rubiginone B2, Antibiotic X-1488E, 6-Hydroxytetrangulol, and Tetrangulol. Tetrahedron. 2000;56:10249–54. https://doi.org/10.1016/S0040-4020(00)00869-3.
Maehr H, Liu CM, Liu M, Perrotta A, Smallheer JM, Williams TH, Blount JF. Five Novel Metabolites Related to Benz[a]Anthracene from an Unidentified Actinomycete Designated x-14881. J Antibiot (Tokyo). 1982;35:1627–31. https://doi.org/10.7164/antibiotics.35.1627.
Speitling M, Nattewan P, Yazawa K, Mikami Y, Grün-Wollny I, Ritzau M, Laatsch H, Gräfe U. Demethyl Mutactimycins, New Anthracycline Antibiotics from Nocardia and Streptomyces Strains. J Antibiot (Tokyo). 1998;51:693–8. https://doi.org/10.7164/antibiotics.51.693.
Kunnari T, Tuikkanen J, Hautala A, Hakala J, Ylihonko K, Mäntsälä P. Isolation and Characterization of and 8-Demethoxy Steffimycins and Generation of 2,8-Demethoxy Steffimycins in Streptomyces Steffisburgensis by the Nogalamycin Biosynthesis Genes. J Antibiot (Tokyo). 1997;50:496–501. https://doi.org/10.7164/antibiotics.50.496.
Izumikawa M, Kawahara T, Hwang-Hwan J, Takagi M, Shin-Ya K. JBIR-107, a New Metabolite from the Marine-Sponge-Derived Actinomycete, Streptomyces Tateyamensis NBRC 105047. Biosci Biotechnol Biochem. 2013;77:663–5. https://doi.org/10.1271/bbb.120832.
Kunz AL, Labes A, Wiese J, Bruhn T, Bringmann G, Imhoff JF. Nature’s Lab for Derivatization: New and Revised Structures of a Variety of Streptophenazines Produced by a Sponge-Derived Streptomyces Strain. Mar Drugs. 2014;12:1699–714. https://doi.org/10.3390/md12041699.
Harada KI, Nishida F, Takagi H, Suzuki M, Iwashita T. Studies on an Antibiotic, Albocycline VII.1) Minor Components of Albocycline. J Antibiot (Tokyo). 1984;37:1187–97. https://doi.org/10.7164/antibiotics.37.1187.
Singh S, Samanta TB. A121–an Antifungal Compound from Streptomyces Species. Microbios. 1992;71:217–24.
Alhadrami HA, Sayed AM, El-Gendy AO, Shamikh YI, Gaber Y, Bakeer W, Sheirf NH, Attia EZ, Shaban GM, Khalifa BA, et al. A Metabolomic Approach to Target Antimalarial Metabolites in the Artemisia Annua Fungal Endophytes. Sci Rep. 2021;11:1–11. https://doi.org/10.1038/s41598-021-82201-8.
Bhattarai, B.R.; Khadayat, K.; Aryal, N.; Aryal, B.; Lamichhane, U.; Bhattarai, K.; Rana, N.; Regmi, B.P.; Adhikari, A.; Thapa, S.; et al. Untargeted Metabolomics of Streptomyces Species Isolated from Soils of Nepal. Processes 2022, 10, https://doi.org/10.3390/pr10061173.
Raheem DJ, Tawfike AF, Abdelmohsen UR, Edrada-Ebel RA, Fitzsimmons-Thoss V. Application of Metabolomics and Molecular Networking in Investigating the Chemical Profile and Antitrypanosomal Activity of British Bluebells (Hyacinthoides Non-Scripta). Sci Rep. 2019;9:1–13. https://doi.org/10.1038/s41598-019-38940-w.
Tawfike AF, Romli M, Clements C, Abbott G, Young L, Schumacher M, Edrada-Ebel R, Diederich M, Farag M, Edrada-Ebel R. Isolation of Anticancer and Anti-Trypanosome Secondary Metabolites from the Endophytic Fungus Aspergillus Flocculus via Bioactivity Guided Isolation and MS Based Metabolomics. J Chromatogr B. 2019;1106:71–83. https://doi.org/10.1016/j.jchromb.2018.12.032.
Khan S, Garg A, Camacho N, Van Rooyen J, Kumar Pole A, Belrhali H, De RibasPouplana L, Sharma V, Sharma A. Structural Analysis of Malaria-Parasite Lysyl-TRNA Synthetase Provides a Platform for Drug Development. Acta Crystallogr Sect D Biol Crystallogr. 2013;69:785–95. https://doi.org/10.1107/S0907444913001923.
Freist W, Gauss DH. Lysyl-TRNA Synthetase. Biol Chem. 1995;376:451–72.
Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, McCormack SL, Plouffe DM, Meister S, Schuierer S, et al. Selective and Specific Inhibition of the Plasmodium Falciparum Lysyl-TRNA Synthetase by the Fungal Secondary Metabolite Cladosporin. Cell Host Microbe. 2012;11:654–63. https://doi.org/10.1016/j.chom.2012.04.015.
Lessmann H, Krupa J, Lackner H, Jones PG. Neue Juglomycine Zeitschrift für Naturforsch B. 1989;44:353–63.
Maskey RP, Lessmann H, Fotso S, Grün-Wollny I, Lackner H, Laatsch H. Juglomycins G-J: Isolation from Streptomycetes and Structure Elucidation. Zeitschrift fur Naturforsch Sect B J Chem Sci. 2005;60:183–8. https://doi.org/10.1515/znb-2005-0210.
Ahmad T, Arora P, Nalli Y, Ali A, Riyaz-Ul-Hassan S. Antibacterial Potential of Juglomycin A Isolated from Streptomyces Achromogenes, an Endophyte of Crocus Sativus Linn. J Appl Microbiol. 2020;128:1366–77.
Yazawa K, Takahashi K, Mikami Y, Arai T, Saito N, Kubo A. Isolation and Structural Elucidation of New Saframycins Y3, Yd-1, Yd-2, Ad-1, Y2b and Y2b-D. J Antibiot (Tokyo). 1986;39:1639–50. https://doi.org/10.7164/antibiotics.39.1639.
Arai T, Yazawa K, Takahashi K, Maeda A, Mikami Y. Directed Biosynthesis of New Saframycin Derivatives with Resting Cells of Streptomyces Lavendulae. Antimicrob Agents Chemother. 1985;28:5–11.
Acknowledgements
Not applicable.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Conceptualization, U.R.A, H.S.B.., H.M.H., and E.M.O.; methodology, U.R.A, N.M., M.D., and T.D.; Software, N.M., and M.D.; supervision, U.R.A, N.M.G.; visualization, H.S.B., E.M.O., H.M.H. and U.R.A; data curation, U.R.A and H.S.B.; writing, original draft preparation, H.S.B.; writing—review and editing, U.R.A, N.M.G., H.S.B., N.A., M.D., H.M.H., T.D., and E.M.O. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure (S1). LC/HRMS-negative mode total ion chromatogram of M1 solid extract (M1-S). Figure (S2). LC/HRMS-positive mode total ion chromatogram of M1 solid extract (M1-S). Figure (S3). LC/HRMS-positive mode total ion chromatogram of ISP2 solid extract (ISP2-S). Figure (S4). LC/HRMS-negative mode total ion chromatogram of ISP2 solid extract (ISP2-S). Figure (S5). LC/HRMS-negative mode total ion chromatogram of Oligo solid extract (Oligo-S). Figure (S6). LC/HRMS-positive mode total ion chromatogram of Oligo solid extract (Oligo-S). Figure (S7). LC/HRMS-negative mode total ion chromatogram of M1 liquid extract (M1-L). Figure (S8). LC/HRMS-positive mode total ion chromatogram of the M1 liquid extract (M1-L). Figure (S9). LC/HRMS-negative mode total ion chromatogram of ISP2 liquid extract (ISP2-L). Figure (S10). LC/HRMS-positive mode total ion chromatogram of ISP2 liquid extract (ISP2-L). Figure (S11). LC/HRMS-positive mode total ion chromatogram of Oligo liquid extract (Oligo-L). Figure (S12). LC/HRMS-negative mode total ion chromatogram of Oligo liquid extract (Oligo-L).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gamaleldin, N.M., Bahr, H.S., Millán-Aguiñaga, N. et al. Targeting antimalarial metabolites from the actinomycetes associated with the Red Sea sponge Callyspongia siphonella using a metabolomic method. BMC Microbiol 23, 396 (2023). https://doi.org/10.1186/s12866-023-03094-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-03094-3