
Abstract
In this study, 25 actinomyces isolates were obtained from 10 different poultry farms and tested for their keratinase activity. The isolate with the highest keratinase activity was identified through molecular identification by PCR and sequencing of the 16S rRNA gene to be Streptomyces spp. and was named Streptomyces werraensis KN23 with an accession number of OK086273 in the NCBI database. Sequential mutagenesis was then applied to this strain using UV, H2O2, and SA, resulting in several mutants. The best keratinolytic efficiency mutant was designated as SA-27 and exhibited a keratinase activity of 106.92 U/ml. To optimize the keratinase expression of mutant SA-27, the Response Surface Methodology was applied using different parameters such as incubation time, pH, carbon, and nitrogen sources. The optimized culture conditions resulted in a maximum keratinase specific activity of 129.60 U/ml. The genetic diversity of Streptomyces werraensis KN23 wild type compared with five mutants was studied using Inter-simple sequence repeat (ISSR). The highest total and polymorphic unique bands were revealed in the S. werraensis KN23 and SA-18 mutant, with 51 and 41 bands, respectively. The dendrogram based on combined molecular data grouped the Streptomyces werraensis and mutants into two clusters. Cluster I included SA-31 only, while cluster II contained two sub-clusters. Sub-cluster one included SA-27, and sub-cluster two included SA-26. The sub-cluster two divided into two sub-sub clusters. Sub-sub cluster one included SA-18, while sub-sub cluster two included one group (SA-14 and S. werraensis KN23).
Similar content being viewed by others
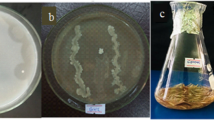


Introduction
One of the most pressing environmental pollution problems is the accumulation of keratinous wastes in slaughterhouses. Feathers, which are produced from poultry processing industries, are a significant by-product, as they make up 5–7% of chicken body weight. It is estimated that worldwide consumption of chicken generates several million tons of feathers annually, with approximately 2 million tons being generated as a by-product of the poultry industry globally [1, 2].
Despite the many applications of feathers, such as decorative applications, medical devices, fertilizer, dusters, bedding materials, and feedstock, a significant amount of feathers are still released into the environment without proper treatment. Traditional techniques of processing feathers, such as chemical processing and stem pressure cooking, may turn feathers into animal feeds, but they consume a large amount of energy and destroy some amino acids. Additionally, feathers keratin has become a source of pollutants due to its recalcitrant nature [3]. To address this issue, many researchers are interested in finding economic methods to convert feathers into value-added products [4]. Numerous studies have shown that different microorganisms can efficiently degrade feathers [5, 6].
Keratinase is an enzyme that can hydrolyze keratin. Diverse groups of microorganisms are known to produce keratinase [7], including bacteria such as Bacillus subtilis [8] and Chryseo bacterium [9], as well as actinomycetes from the Streptomyces genus [10]. Additionally, fungi such as Microsporum, Aspergillus [11], Chrysosporium [12], and Trichoderma [7] are also known to possess active keratinolytic properties. Enzymatic degradation of keratin using the enzymes isolated from bacterial cells or the bacterial culture has been proposed as a clean and inexpensive method of converting feather keratin into single amino acids and polypeptides.
Given the effectiveness of traditional mutagenesis for creating mutants that produce improved yields of several microbial enzymes, such as α-galactosidase and lipase [13], it is reasonable to assume that a similar strategy could be successfully applied to enhance the ability of keratinase-producing strains.
Response Surface Methodology (RSM) is an experimental approach that efficiently determines the optimal conditions for a multivariable approach [14, 15]. RSM is a collection of mathematical and analytical techniques that are used to design experiments, construct models, measure responses, analyze results, and explore relationships between controlled factors under experimental conditions. This methodology is particularly useful in bioprocess optimization for identifying the most favorable conditions.
Inter Simple Sequence Repeat (ISSR) markers are a PCR-based method that do not require any prior genomic information [16]. ISSR markers are highly polymorphic and can be used in studies on genetic diversity, phylogeny, gene tagging, genome map**, and evolutionary biology [17]. Molecular markers like ISSR provide additional tools for characterizing, differentiating, and assessing genetic variation in collections. The ISSR-PCR technique of DNA fingerprinting has been employed in this work. It utilizes arbitrary, multi-loci markers generated by PCR amplification with microsatellite primers, which are quick, cost-effective, and easy to analyze [18]. Due to their high level of polymorphism and reproducibility, this fingerprinting technique is widely used, particularly in plants [17, 19], yeasts [20], and bacteria [21].
Therefore, the objective of this study was to isolate a new keratinase-producing actinomycetes strain and apply sequential mutagenesis to enhance keratinase productivity.
Materials and methods
Collecting samples
A total of 10 poultry waste samples were collected from 10 different farmers in Cairo, Egypt, from a depth of 5–10 cm, in sterile containers. Authors have permission for collection of samples obtained from the farm owners.
Culture media and isolation of keratinase-producing bacteria
The basic medium used for isolation and fermentation of the feather-degrading bacteria was according to [22, 23]. Plate count (P.C) agar medium (Himedia, West Chester, Pennsylvania, USA) was used for actinomycetes growth.
Preparation of keratin solution
Soluble keratin was prepared from chicken feathers obtained from local poultry waste according to the method of [24, 25].
Keratinase production and enzyme assay
A pure single colony of freshly selected actinomycetes isolate, which was grown on P.C agar medium, was aseptically transferred for keratinase production using fermentation medium according to [23]. After incubation, the supernatant containing the enzyme excreted was used in quantitative keratinase assay. Keratinase enzyme activity was measured using keratin solution as a substrate. In brief, 1.0 ml of cell-free supernatant (crude enzyme) was incubated with 1 ml keratin in 0.05 M Tris–HCl buffer (pH 8.0) and incubated at 50 °C for 10 min as reported by [26]. One unit (U/ml) of keratinolytic activity was defined as an increase of corrected absorbance of 280 nm (A280) with the control for 0.01 per minute under the conditions described above and calculated by the following equation: U = 4 × n × A280/(0.01 × 10), where n is the dilution rate; 4 is the final reaction volume (ml); 10 is the incubation time (min).
Determination of protein content and residual hydrolysates
Protein content was determined as described by [27] using bovine serum albumin as a standard. The residual hydrolysates were composed of undigested feathers and cells. The residual hydrolysate’s weight was determined according to [23].
Molecular identification of keratinolytic actinomyces
DNA extraction: The extraction method was applied according to the manufacturer of (Applied Biotechnology Co., Egypt), the actinomyces isolate was identified based on partial sequencing of the 16S rRNA gene, using the universal primers as follow, forward primer 63f (5’- CAGGCCTAACACATGCAAGTC-3’) and reverse primer 1387R (5’- GGGCGGWGTGTACAAGGC-3’). PCR product was purified and sequenced as described previously [28]. PCR program was 95 °C for 3 min, 30 cycles of denaturation at 95 °C for 45 s, annealing at 56 °C for 45 s, and extension at 72 °C for 1 min /1kbp, final extension 72 °C for 5 min. PCR product was purified using MEGA Quick-Spin total fragment DNA purification kit as instructed by the manufacturer. Sequencing was performed by the Sanger method [29].The phylogenetic tree was constructed using MAFFT alignment [30]. https://mafft.cbrc.jp/alignment/server/phylogeny.html.
Mutagenesis
The wild-type S. werraensis KN23was cultured on P.C broth medium at 37 °C for 2-5 days. Then, 10 ml of culture were centrifuged at 9000 g at 4 °C for 10 min to separate the cell biomass. Then, the cell biomass pellet was resuspended in 10 ml of sterilized saline (0.9%).
For UV-induced mutagenesis [Full size image
Adequacy of the model
To ensure the accuracy of the model, a random set of 30 production combinations was used to experimentally retest the keratinase production [44, 46]. The optimized conditions determined from the model predicted a keratinase production of 129.60 U/ml. The experimental value obtained was 122.54 U/ml, which was close to the predicted value. The model was found to be valid and reliable, as confirmed by the close agreement between the experimental and predicted values, as indicated in Table 4.
Factor coding is Coded.
Sum of squares is Type III – Partial.
The Model F-value of 21.05 indicates that the model is significant, and there is only a 0.01% chance that such a large F-value could occur due to noise.
Model terms with P-values less than 0.0500 are considered significant, and in this case, A, B, AC, and A2 are significant model terms. Model reduction may improve the model if there are many insignificant model terms (excluding those required to support hierarchy), with values greater than 0.1000.
The Lack of Fit F-value of 1.34 implies that the Lack of Fit is not significant relative to the pure error, and there is a 39.24% chance that a Lack of Fit F-value this large could occur due to noise. Non-significant Lack of Fit is desirable as it means that the model fits well.
The Predicted R2 of 0.7798 is in reasonable agreement with the Adjusted R2 of 0.9063; the difference is less than 0.2.
The Adeq Precision measures the signal to noise ratio, and a ratio greater than 4 is desirable. The ratio of 14.967 indicates an adequate signal, and this model can be used to navigate the design space.
Select the highest order polynomial where the additional terms are significant and the model is not aliased.
Molecular depiction
Molecular description of S.werraensis KN23 and its mutants were conducted using ISSR primers (ISSR-152, ISSR-153, ISSR-154, UBC851, ISSR-157, ISSR-158, and ISSR-861). The ISSR analysis produced a total of 122 markers, with 112 of them being polymorphic, and unique bands displaying 91% polymorphism, as shown in Table 8 and Fig. 7. The highest number of total bands were displayed in S. werraensis KN23 and S. A-18, with 51 bands, followed by the mutant S. A-27 with 50 bands, as indicated in Table 9 and Fig. 6. The number of polymorphic bands with unique markers was 41 for KN23 and SA-18, whereas the mutants S.A-27, S.A-31, and S.A-14 showed 40, 39, and 37 bands, respectively. The lowest number of total bands and polymorphic bands with unique markers were displayed in isolate SA-26, with 39 and 29 bands, respectively, compared to the other genotypes, as shown in Tables 9 and 10, and Figs. 6, 7 and 8.

The relationship between total bands of the different primers used for the detection of the strain KN23 strain and five mutants

The relationship between, polymorphic with unique bands and polymorphism percentage of the seven ISSR primers used for the detection of six Streptomyces werraensis KN23 and five mutants

The Inter-Simple Sequence Repeats (ISSR) amplification pattern obtained for six samples of Streptomyces werraensis strain KN23 and five mutants, a primer ISSR-152, b primer ISSR-153, c primer ISSR-154, d primer UBC-851, e primer ISSR-157 f primer ISSR-158 and g primer ISSR-861
Proximity matrix analysis (Genetic Similarity)
Data showed in Table 11 recorded (15) pairwise comparisons to debate the genetic relationships among 6 Streptomyces werraensis genotypes detected in terms of similarity. The genetic similarity ranged from (0.380 to 0.571) with an average of (0.475), where the biggest value of genetic similarity was (0.571) among (S.werraensisKN23 and SA-14) and the lowest value of similarity was (0.380) between (SA-18 and SA-31), respectively. Also, high genetic similarity values were observed within, (SA-14 and SA-18) (0.551), (S.werraensis KN23 and SA-18) (0.549), (SA-26 and SA-27) (0.539),While (SA-14 and SA-26) (SA-18 and SA-27) was (0.535) on the other hand (S.werraensis KN23 and SA-26) (0.489) (S.werraensisKN23 and SA-27) (0.475) (KN23 and SA-31) (0.420) finaly the (SA-14 and SA-31) (0.396), (SA-14 and SA-27) (0.392) and (SA-18 and SA-31) (0.380) respectively.
Cluster analysis (Phylogenetic tree)
Results of cluster analysis viewed in Fig. 9 divided all Streptomyces werraensis genotypes into two main clusters. The cluster I included (SA-31) only while, cluster II contained two sub-clusters. The sub-cluster one included (SA-27), the sub-cluster two included (SA-26). While, the sub-cluster two divided into two sub-sub clusters. The sub-sub cluster one included (SA-18), while the sub-sub cluster two included one group (SA-14 and S.werraensis KN23).

UPGMA based dendrogram showing the genetic relationship among 6 genotypes of S. werraensisusing 7 primers of ISSR
