
Abstract
Background
Ampelopsideae J. Wen & Z.L. Nie is a small-sized tribe of Vitaceae Juss., including ca. 47 species from four genera showing a disjunct distribution worldwide across all the continents except Antarctica. There are numerous species from the tribe that are commonly used as medicinal plants with immune-modulating, antimicrobial, and anti-hypertensive properties. The tribe is usually recognized into three clades, i.e., Ampelopsis Michx., Nekemias Raf., and the Southern Hemisphere clade. However, the relationships of the three clades differ greatly between the nuclear and the plastid topologies. There has been limited exploration of the chloroplast phylogenetic relationships within Ampelopsideae, and studies on the chloroplast genome structure of this tribe are only available for a few individuals. In this study, we aimed to investigate the evolutionary characteristics of plastid genomes of the tribe, including their genome structure and evolutionary insights.
Results
We sequenced, assembled, and annotated plastid genomes of 36 species from the tribe and related taxa in the family. Three main clades were recognized within Ampelopsideae, corresponding to Ampelopsis, Nekemias, and the Southern Hemisphere lineage, respectively, and all with 100% bootstrap supports. The genome sequences and content of the tribe are highly conserved. However, comparative analyses suggested that the plastomes of Nekemias demonstrate a contraction in the large single copy region and an expansion in the inverted repeat region, and possess a high number of forward and palindromic repeat sequences distinct from both Ampelopsis and the Southern Hemisphere taxa.
Conclusions
Our results highlighted plastome variations in genome length, expansion or contraction of the inverted repeat region, codon usage bias, and repeat sequences, are corresponding to the three lineages of the tribe, which probably faced with different environmental selection pressures and evolutionary history. This study provides valuable insights into understanding the evolutionary patterns of plastid genomes within the Ampelopsideae of Vitaceae.
Similar content being viewed by others

Introduction
Ampelopsideae is a small-sized tribe and the first diverged lineage of the grape family of Vitaceae, including ca. 47 species from four genera showing a disjunct distribution worldwide across all the continents except Antarctica [1, 2]. Members of the tribe are morphologically characterized by the inflorescence mostly five-parted with a cup-shaped disc, which is slightly lobed and adnate proximally to the base of the ovary while being free distally [2]. The tribe contains many species that can be used medicinally, such as Ampelopsis delavayana Planch. ex Franch. and Ampelopsis japonica (Thunb.) Makino. possess immunomodulatory and antimicrobial activity, and treats hypertension function [3,4,5].
Three lineages are usually recognized within Ampelopsideae, i.e., Ampelopsis, Nekemias, and the Southern Hemisphere clade [1, 6, 7]. However, the relationships of the three clades differ greatly between the nuclear and the plastid topologies [1, 7, 8]. Nuclear data indicate that Ampelopsis is the first diverged lineage, sister to a clade including Nekemias and the clade composed of Rhoicissus Planch. and Clematicissus Planch. from the Southern Hemisphere [7, 8]. Nonetheless, the plastid tree proposed Nekemias as the first diverged lineage within the tribe [7]. Nekemias is similar in distribution as Ampelopsis, with most species occurring in East Asia and only a few in North America [1]. Because of their distributional and morphological similarities, taxonomists traditionally placed Nekemias in Ampelopsis [9]. Both nuclear and plastid gene data support the embedding of taxa from Southern Hemisphere into the traditional Ampelopsis [1, 8].
Plastids genomes in angiosperms are highly conserved with similar structure, gene sequences and organization, with length between 120 to 160 kb in size [10]. They comprise a large single copy region (LSC; 80–90 kb), a small single copy region (SSC; 16–27 kb), and two inverted repeat regions (IRs) of approximately 20–28 kb each [10]. Because of their conserved structure, low occurrence of recombination, and primarily uniparental inheritance, plastid sequences have been extensively employed as preferred markers for plant phylogenetics and evolution [1, 11,12,13,14]. Although the plastid genome is usually conserved [15, 16], structural rearrangements, gene loss, IR expansions, and inversions occur in certain lineages and provide useful insights into phylogenetic evolution in plants [17, 18]. For example, plastid genome sequences have been utilized for DNA barcoding, phylogenetic, transplastomic and population questions [19,20,21,22,23,24].
Recent advances in genomic sequencing have led to the availability of complete plastid genomes, which provide more comprehensive information for phylogenetic studies. Although previous studies have investigated the chloroplast genomes of individual or a few species in the grape family, including some taxa from Ampelopsideae, such as A. delavayana, A. japonica and Nekemias cantoniensis (Hook. & Arn.) J. Wen & Z.L. Nie [25, 26], expanding the sampling of the tribe would be beneficial for understanding the plastid structural evolution within Ampelopsideae.
In this study, we aimed to newly sequence and assemble plastid genomes of a total of 36 species from Ampelopsideae and closely related taxa, in order to investigate the evolutionary characteristics of plastid genomes of the tribe, including their genome structure and evolutionary insights. We hypothesized that a broad sampling of the tribe would provide a more comprehensive understanding of its plastome evolutionary pattern. Our results may also provide insights into the evolution of other taxa of the economically highly significant grape family and inform future research on their molecular, morphological, geographic, and ecological diversification.
Materials and methods
Plant materials, DNA extraction and sequencing
In this study, we sampled a total of 36 accessions, including 30 individuals representing 22 species from Ampelopsideae, plus 6 from other genera of the family (i.e., Parthenocissus Planch., Cissus L., Cayratia Juss. and Pseudocayratia J. Wen, L.M. Lu & Z.D. Chen). All the samples were newly sequenced except that two species from Ampelopsis were obtained from NCBI (MK574541 and MK574542). In accordance with previous researches [1, 7, 8], Leea guineensis G. Don. (MW592489), a species from Leeaceae Dumort., the sister family of Vitaceae, was utilized as a remote outgroup for reconstructing the phylogenetic tree. Information on the plant material (collection localities and voucher specimen numbers) and the associated GenBank accessions are listed in Supplementary Table 1.
A modified CTAB method was used to extract total DNA from either silica gel-dried leaves or plant specimens [27, 28]. Extracted DNAs were quantified on a Qubit 4.0 fluorometer (Thermo Fisher Scientific) using a high-sensitivity kit and then sheared to a target size ca. 300–500 bp by sonication (QSonica Q800RS). DNA libraries were generated with the NEBNext Ultra DNA Kit following the manufacturer’s protocol. The libraries were then sequenced on an Illumina HiSeq 4000 platform using a 150 paired-end protocol.
Data assembly and annotation
Clean raw data were used to assemble complete plastid genome sequences by the program GetOrganelle [29], and then annotated using GeSeq (https://chlorobox.mpimpgolm.mpg.de/geseq.html) [30]. The obtained sequences were checked and manually adjusted in the program Geneious-9.0.2 using Ampelopsis humulifolia Bunge. as a reference. Finally, all the newly sequenced plastid genomes were uploaded to NCBI (Supplementary Table 1). Additionally, plastid genomic maps were generated from https://chlorobox.mpimp-golm.mpg.de/OGDraw.html [31].
Phylogenetic analysis
The completed plastids genome sequences were aligned using MAFFT 7.427 [32]. Phylogenetic analysis was conducted based on maximum likelihood (ML) analysis using the GTRGAMMA nucleotide substitution model with the default parameters in RAxML 7.2.6 [33]. RAxML allows for only a single evolutionary model in partitioned analyses, which was selected according to PartitionFinder2 results. Bootstrap supports (BS) were estimated using a rapid bootstrap** algorithm and 1000 replicates in RAxML.
Plastome comparative analyses
The simple sequence repeats (SSR) were detected by MISA (https://webblast.ipk-gatersleben.de/misa/), with parameters set to ten, five, and four repeats for mononucleotide, dinucleotide, and trinucleotide [34]. Three repeats were used for tetranucleotide, pentanucleotide and hexanucleotide. We used REPuter to analyze forward, palindrome, reverse and complementary sequences with a minimum repeat length of 16 bp and minimum sequence identity greater than 90% [35].
The expansion and contraction of the IR regions were examined with the IRscope (https://irscope.shinyapps.io/irapp/) [36]. The codon usage was analyzed with CodonW [37]. For the nucleotide diversity analysis, complete plastid genome sequences were aligned with MAFFT [32]. A sliding window analysis of window length of 600 bp and step size of 200 bp was used in the DnaSP to estimate the nucleotide diversity values [38]. Structural changes across plastid genomes of Ampelopsideae were analyzed via whole-genome alignment in Mauve 2.4.0 using default parameters [39].
To evaluate the selection pressure on protein-coding genes, we extracted the shared non-redundant genes among species, in which each gene’s CDS-pair of one-by-one species’ combination were extracted and aligned by MAFFT [32]. The rates of synonymous substitutions (Ks) and non-synonymous substitutions (Ka) and Ka/Ks were then calculated by KaKs_Calculator in ParaAT 2.0 [40] using “ParaAT.pl -c 11 -h homologs.txt -n CDS -a PEP -p proc -o OUT -k -f axt -m mafft -v”. The Ka/Ks ratio defines the degree of gene divergence and whether selection pressure is positive (Ka/Ks > 1), purifying (Ka/Ks < 1, particularly if it is less than 0.5), or neutral (Ka/Ks = 1) [41], which is useful for understanding the evolution of protein-coding genes and adaptive developments in species [41, 42].
Results
Basic characteristics of plastid genomes of the tribe
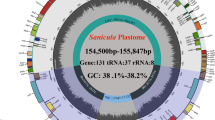
Diagrams of the plastid genomes were presented in Fig. 1. All the plastomes of the tribe show a typical quadripartite structure comprising a LSC region (85,420—93,530 bp) and a SSC region (18,439—21,778 bp) separated by two IR regions (25,689—27,412 bp) (Fig. 1; Table 1). The average GC content of all sequences is ~ 37.4%, including 35.38% for the LSC, 31.85% for the SSC, and 42.6% for the IR region (Table 1). The total number of annotated genes is 133 to 134, comprising 88 to 89 protein-coding, 36 to 37 tRNA, and 8 rRNA genes (Table 1).

The chloroplast genome maps of Ampelopsideae. Transcriptional directions are represented on the circle’s inside (clockwise) and outside (counterclockwise). Genes are color-coded according to their functional groups
Of the 18 duplicated genes in the IR, seven are protein-coding, seven are tRNA, and four are rRNA genes. We observed gene duplication and loss in the plastid genes of some species (Table 2). For example, copies of the rps19 gene were found in all genera of Nekemias. Additionally, Nekemias arborea (L.) J. Wen & Boggan has a deletion of the ycf1. We also found pseudogenes in our assembled data, such as rps19 pseudogene (ψrps19), ycf1 pseudogene (ψycf1), and ndhI pseudogene (ψndhI) (Table 2).
Phylogenetic relationships
A ML tree was reconstructed based on all the plastid genome data (Fig. 2). The plastid phylogeny of Ampelopsideae has a high level of resolution as most relationships supported with strong to medium support values (BS > 75%). Three main clades were recognized within the tribe, corresponding to Ampelopsis, Nekemias, and the Southern Hemisphere clade, respectively, and all received 100% bootstrap values (Fig. 2). Within the Ampelopsis clade, the North American species Ampelopsis cordata Michx. represents as the first diverged lineage, sister to the remaining members from East Asia (Fig. 2). The North American species from Nekemias serves as the first divergent lineage sister to the East Asian group (Fig. 2). For taxa from the Southern Hemisphere, the African Rhoicissus is sister to the expanded Clematicissus with taxa from the South American species forming a clade sister to the Australian species (Fig. 2).

A ML tree of Ampelopsideae inferred from complete chloroplast genomes. Numbers near nodes represent bootstrap support values. The heat map shows different repeat sequence types and numbers for each taxon
Plastome structure and length variation
The variation in total length of the chloroplast genomes and the sizes of each region among the three clades within the tribe are presented in Fig. 3. The plastid sequences of the tribe exhibit large variation in length, ranging from 160,692 to 163,219 bp (Table 1). Nekemias shows the longest average length of 162,854 bp within the tribe, ranging from 162,165 to 164,115 bp, and Ampelopsis has a close average length of 162,233 bp, ranging from 161,430 to 162,468 bp (Table 1). In contrast, the Southern Hemisphere lineage exhibits the shortest average length of 161,106 bp, ranging from 160,389 to 162,432 bp (Table 1). The LSC region of Ampelopsis is the largest, with an average size of 90,184 bp (ranging from 89,627 bp to 90,419 bp), and Nekemias shows the next largest average length as 89,391 bp (ranging from 88,868 bp to 90,959 bp) (Table 1). The IR region of Nekemias has an average size of 27,109 bp (ranging from 25,689 bp to 27,412 bp), while the other two clades have similar smaller average size (Table 1). The SSC region is relatively similar among the three clades, ranging from 18,895 to 21,778 bp.

Length variation in the plastid genomes of Ampelopsideae. The y-axis values are minus data for the smallest genome of Pseudocayratia dichromocarpa
The plastid genomes of Ampelopsideae show no significant differences in the boundaries of the IR and SSC regions, except for N. arborea, where the ycf1 pseudogene and ycf1 gene are not found at the IRb-SSC boundary and the IRa-SSC boundary, respectively (Fig. 4). Additionally, in Rhoicissus digitata (L.f.) Gilg & Brandt, the ndhF gene spanned 42 bp across the JSB (IRb-SSC boundary) (Fig. 4). In the LSC and IR boundaries, Ampelopsis and the Southern Hemisphere taxa show the rps19 gene spanning the JLB (LSC-IRb boundary), the rpl22 gene located near the JLB in the LSC region, and the rpl2 gene on the left side of JLA (Fig. 4). In contrast, Nekemias species exhibit different pattern, with the rpl22 gene presented on the JLB, the rps19 gene located in the IR region to the right of JLB, and the rps19 gene on the left side of JLA (Fig. 4).

Comparison of the gene order and IR/SC junction sites in Ampelopsideae plastomes (covering the three lineages of the tribe). The number of base pairs indicates the distance between the end of the gene and the junction site. Boxes above and below the regions represent genes transcribed in the forward and reverse DNA strands, respectively
No gene rearrangements were found in the plastid genome of each genus in the Ampelopsideae (Fig. S1). Furthermore, the gene arrangement of the tribe was found to be similar to other species of Vitaceae (Fig. S1). Nucleotide diversity values were calculated for Ampelopsideae, with the highest nucleotide diversity observed in the SSC region (0.0423) and those in the IR region were less than 0.003 (Fig. S2).
Repetitive sequences and SSR
Among the four different types of repetitive sequences, the number of forward repeats (F-type) and palindromic repeats (P-type) is higher than the number of complement repeats (Fig. 2). N. cantoniensis 2 has the largest total number of 602 repetitive sequences (Fig. 2). However, the number of the two types varied widely among species, with the largest number found from N. cantoniensis 2, including 261 P-types and 263 F-types (Fig. 2). In addition, the number of duplication of both F-type and P-type was relatively high in Nekemias, while the number of reverse repeats (R-type) was slightly lower than that of other genera (Fig. 2).
A total of 27 different types of SSRs were found in the tribe (Fig. 5). A/T and AT/AT repetitions account for most of them, and A/T, AT/AT, AAT/ATT and AAAT/ATTT are the simple repetition type common to all species. Some simple repeat types such as ACT/AGT, AAAC/GTTT, and AATT/AATT occur once in some species, while AAAG/CTTT and AATC/ATTG are missing in some species (Table S2). The number and type of SSRs in the SC and IR regions are different. Most of the SSRs were found in the SC region, with more than 75% of the total SSRs found in the LSC region.

SSR types in Ampelopsideae plastomes
Codon usage
The relative synonymous codon usage (RSCU) frequency was calculated using 88 protein-coding sequences from the plastid genome. Among all amino acids, Leu is the mostly used codons and Cys is the least one (Fig. S3). Compared synonymous codon usage analysis (Fig. S3, Fig. 6) discovered that RSCU value of 30 to 31 codons is greater than 1 (Fig. 6). Met and Trp have no biased usage (RSCU = 1). Among the codons with RSCU > 1 in the Ampelopsideae, only the Leu codon (UUG) is G-ending, and the other 29 to30 codons are A or U-ending. AGA, which encodes the Arg amino acid, is the most preferred codon (minimum value of preference index > 1.756), while CGC is the one with the lowest preference index (maximum value of preference index < 0.377). In the cluster analysis using codon preference, the clustering tree was largely grouped into three major blocks, corresponding to the three clades of the tribe (Fig. 6).

The heat map of codon usage bias in the chloroplast genomes of Ampelopsideae. The color depth represents the Euclidean distance
Selective pressure evaluation
We compiled a data matrix comprising the Ka/Ks values of 54 gene pairs across a total of 35 species (Fig. 7). Excluding genes for which the Ka/Ks values could not be determined, our analysis yielded a total of 2,240 gene loci with Ka/Ks less than 0.5, 218 gene loci with Ka/Ks greater than 0.5 but less than 1, and only 58 gene loci exhibited with Ka/Ks greater than 1 (Fig. 7). We detected that 34 genes in Ampelopsideae exhibited Ka/Ks values close to or equal to 0 (Fig. 7). Additionally, we observed positive selection acting on the psaI gene across the whole Ampelopsideae, while ccsA, cemA, psbK, rpl32, and ycf2 exhibit Ka/Ks greater than 1 in some species. The rpl32 gene is under positive selection in almost all members of Nekemias and some Clematicissus species from the Southern Hemisphere (Fig. 7).

The heat map showing pairwise Ka/Ks ratios between concatenated single-copy coding sequences among Ampelopsideae plastid genomes
Disussions
Plastid genomes tend to be stable and conserved in plants [43, 44]. Our findings (Table 1) suggested that Ampelopsideae plastomes are largely consistent with previous reports in terms of structure, gene number, RNA and protein-coding genes, and GC content [45,46,47,48]. The GC content was found to be higher in the IR region than the SC region, likely due to the agglomeration of four rRNA genes in the IR region [49]. Conversely, the SSC region exhibited a lower GC content than that of the LSC region, which could be attributed to ndh gene clustering in the SSC region.
This study reconstructed well-supported phylogenetic relationships of the Ampelopsideae based on the plastid genomic sequences, which represented the first phylogeny of the tribe based on a broad sampling of plastid genomes (Fig. 2). Our study was largely consistent with previous results [2, 7, 11]. The plastid genomes provided robust support for the recently resurrected Nekemias as a distinct monophyletic genus, separate from Ampelopsis [7,8,9, 50], suggesting Nekemias as the first diverged lineage within the tribe, sister to a clade including Ampelopsis and taxa from the Southern Hemisphere, a backbone relationship of the tribe congruent with those reported by recent studies [1, 7, 11, 50, 51]. Furthermore, our data improved resolution throughout the tribe compared with previous studies, with almost all nodes being strongly supported (Fig. 2).
Ampelopsideae exhibits variations in size across the plastid genome as well as within the LSC and IR regions consistent with the phylogenetic relationships (Fig. 3). Most species in Ampelopsideae show minimal variation in the SSC (Fig. 3), indicating that the impact on plastid genome size is primarily driven by changes in the LSC and IR regions. The length variation of the IR regions is commonly found in the plastid genomes of angiosperms, which often leads to changes the number of genes in various plant lineages [43, 48, 52,53,54,55,56]. Compared with the other two clades, the Nekemias clade shows a distinguishable expansion in the IR region (Fig. 3). Nekemias has a complete duplicated copy of the rps19 gene in the IRa region and the rpl22 gene is incorporated more often into the IRb region (Fig. 4). Correspondingly, the LSC region is reduced in Nekemias due to the rps19 gene is assigned to the IR region. The IR expansion resulted from the generation of a pseudo-copy or functional gene copy of a single-copy gene with transferring from LSC or SSC to IRs [48, 56, 57]. Previous reports have shown that in monocots that IR expansion occurs at the IRa/LSC boundary, resulting in a duplicated copy of the trnH-GUG gene adjacent to rps19 at the IRb/LSC boundary [17]. The rps19 protein is a component of the 40S ribosomal subunit and belongs to a family of ribosomal proteins restricted to eukaryotes and archaea [58]. Although the evolutionary significance is unclear for the increased copy of the rps19 in Nekemias, we illustrated an interesting case of the independent duplication of rps19 in the IR region of Nekemias within the Ampelopsideae.
On the other hand, the overall length of the plastid genome and LSC region for the Southern Hemisphere clade is smaller than those of the other two clades (Fig. 3). Although the LSC region has expanded into the IR region, it did not ultimately result in an overall expansion of the LSC region. This suggests that the expansion of the LSC region into the IR region is not the primary cause of the size variations in different regions for the Southern Hemisphere clade. Because there is no gene loss in the LSC region, the size variations in this clade are probably due to partial deletions and insertions in intergenic spacer regions. Furthermore, the expansion and contraction of the SSC region specific to N. arborea and Nekemias grossedentata 1 (Hand.-Mazz.) J. Wen & Z.L. Nie (Fig. 3) may be the result of interactions with the IR region and the loss of intergenic region segments within the SSC region.
Repetitive sequences play diverse roles in cellular processes, including gene evolution, gene expression, mRNA stabilization, gene organization, gene mobility, cellular immunity against foreign genes, and even gene engineering in prokaryotes and eukaryotes [59,60,61,62,63,64,65]. The F-type and P-type are more abundant than R-type and C-type, a pattern consistent with previous findings in other plant taxa [66, 67]. These repeat sequences are pivotal for genome reconfiguration and have been associated with numerous insertions and deletions [57]. The prevalence of such repeats could enhance nucleotide diversity [68], providing a basis for evolutionary and population genetic studies [69]. This could signify the important roles that F-type and P-type in genetic recombination, DNA repair, and replication fidelity. The SSRs in Ampelopsideae, particularly the highly abundant in poly-A and T motifs (Table S2), are potential molecular markers due to their high polymorphism and mutation rates [25, 70,71,72,73,74,75].
The types and content of repetitive sequences in the Ampelopsideae display variation among clades (Fig. 2), indicating that they may have undergone distinct evolutionary trajectories and adapted to different ecological niches. Notably, the Nekemias shows a higher abundance of F-type and P-type repeats (Fig. 2), which may suggest that the genus possesses genome characteristics distinct from both Ampelopsis and the Southern Hemisphere taxa. Interestingly, despite both Nekemias and Ampelopsis primarily distributed in East Asia, Ampelopsis shared a lower number of F- and P-type but relatively higher number of R-type and C-type repeats similar to that of the Southern Hemisphere taxa (Fig. 2). Ampelopsis and Nekemias have a similar distribution and habitats mainly in East Asia, these differences of repeat types likely reflect the distinct evolutionary history and ecological adaptations to local niches, which may have arisen in response to different selective pressures and environmental conditions [1, 2]. Overall, the identification and characterization of repetitive sequences in different taxa of the Ampelopsideae provide valuable insights into understanding their evolutionary diversification and ecological adaptation in East Asia.
We found that specific codons are more frequently used in the nucleotide sequences of protein-coding genes in the plastid genome of the Ampelopsideae than other synonymous codons (Fig. S3), consistent with previous reports [25]. All preferred synonymous codons (RSCU > 1) end with A or U, which may contribute to the bias towards A/T bases throughout the genome. In contrast, codons ending with C, such as CGC (Arg), UGC (Cys), CAC (His), and AGC (Ser), have relatively low RSCU values (Fig. S3). RSCU can affect gene expression by regulating the accuracy and efficiency of gene translation, with stronger RSCU leading to higher gene expression levels[76,90]. Notably, the highest nucleotide diversity was found at the boundary between the IRs and SSC regions (Fig. S2), which might be caused by fine rearrangements during the contraction and expansion of the IR region's boundaries.
Purifying selection, one of the most prevalent forms of natural selection, constantly removes deleterious mutations in populations [91]. The low Ka/Ks ratios observed at the chloroplast genome within the tribe indicate that most genes are subject to purifying selection to retain conserved functions (Fig. 7). Positive selection has been found in genes related to photosynthesis in some weakly light-adapted aquatic plants [91]. In most cases, genes related to specific environments are typically assumed to be under positive selection [92]. The psaI gene, encoding a reaction protein complex in Photosystem I of plant chloroplasts, plays a crucial role in photosynthetic pigment reactions [93]. The psaI gene may be a candidate gene for adaptive evolution in response to the specific growth environment of Ampelopsideae species since their small and cree** growth habit under forestry push them in competition for sunlight with taller trees or shrubs.
Conclusions
This study demonstrated the conservation of genome size, gene number, and GC content within the Ampelopsideae, with no major gene rearrangements observed. But our results also indicated that plastomes wihin the tribe vary among three lineages in genome length, expansion or contraction of the inverted repeat region, codon usage bias, and repeat sequences, probably due to different environmental selection pressures and evolutionary histories. Furthermore, some specific genes are under positive selection, such as psaI and rpl32, suggesting that they are significant in the Ampelopsideae evolution. Building on the solid phylogenetic and evolutionary framework established here, future studies with even greater taxonomic and genomic sampling may contribute to a better understanding of the diversification patterns in Ampelopsideae in relation to climatic, biogeographic, and ecological factors.
Availability of data and materials
Chloroplast data for all species in this paper have been uploaded to NCBI and collection information and name coding with species samples are recorded in Supplementary Table 1. Additionally, it includes accession numbers of the reference species downloaded from NCBI.
References
Nie ZL, Sun H, Manchester SR, Meng Y, Luke Q, Wen J. Evolution of the intercontinental disjunctions in six continents in the Ampelopsis clade of the grape family (Vitaceae). BMC Evol Biol. 2012;12(1):1–13.
Wen J, Lu LM, Nie ZL, Liu XQ, Zhang N, Ickert-Bond S, Boggan J, Chen ZD. A new phylogenetic tribal classification of the grape family (Vitaceae). J Syst Evol. 2018;56(4):262–72.
Li HJ, Zhang L, Wang SG. Pharmacological action and clinical application of Radix Ampelopsis. Food Drug. 2007;10:60–2.
Nho KJ, Chun JM, Kim DS, Kim HK. Ampelopsis japonica ethanol extract suppresses migration and invasion in human MDA-MB-231 breast cancer cells. Mol Med Rep. 2015;11(5):3722–8.
Chinese Pharmacopoeia Commission. Chinese pharmacopoeia. China Medical Science Press: Bei**g, China. 2015;1:191–3.
Lu L, Cox CJ, Mathews S, Wang W, Wen J, Chen Z. Optimal data partitioning, multispecies coalescent and Bayesian concordance analyses resolve early divergences of the grape family (Vitaceae). Cladistics. 2018;34(1):57–77.
Ma ZY, Nie ZL, Ren C, Liu XQ, Zimmer EA, Wen J. Phylogenomic relationships and character evolution of the grape family (Vitaceae). Mol Phylogenet Evol. 2021;154:106948.
Wen J, Nie ZL, Soejima A, Meng Y. Phylogeny of Vitaceae based on the nuclear GAI1 gene sequences. Botany. 2007;85(8):731–45.
Wen J, Boggan J, Nie ZL. Synopsis of Nekemias Raf., a segregate genus from Ampelopsis Michx.(Vitaceae) disjunct between eastern/southeastern Asia and eastern North America, with ten new combinations. PhytoKeys. 2014;42:11–9.
Cheng H, Li J, Zhang H, Cai B, Gao Z, Qiao Y, Mi L. The complete chloroplast genome sequence of strawberry (Fragaria×ananassa Duch.) and comparison with related species of Rosaceae. PeerJ. 2017;5:e3919.
Soejima A, Wen J. Phylogenetic analysis of the grape family (Vitaceae) based on three chloroplast markers. Am J Bot. 2006;93(2):278–87.
Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7(1):1–17.
Zhang HJ, Zhang X, Landis JB, Sun YX, Sun J, Kuang TH, Li LJ, Tiamiyu BB, Deng T, Sun H, Wang HC. Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). J Syst Evol. 2022;60:1229–40.
Yan LJ, Zhu ZG, Wang P, Fu CN, Guan XJ, Kear P, Zhang CZ, Zhu GT. Comparative analysis of 343 plastid genomes of Solanum section Petota: Insights into potato diversity, phylogeny, and species discrimination. J Syst Evol. 2023;61:599–612.
Palmer JD. Comparative organization of chloroplast genomes. Annu Rev Genet. 1985;19(1):325–54.
Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17:1–29.
Li B, Zheng Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci Rep. 2018;8(1):9285.
Gonçalves DJ, Jansen RK, Ruhlman TA, Mandel JR. Under the rug: abandoning persistent misconceptions that obfuscate organelle evolution. Mol Phylogenet Evol. 2020;151:106903.
Provan J, Powell W, Hollingsworth PM. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol. 2001;16(3):142–7.
Ravi V, Khurana JP, Tyagi AK, Khurana P. An update on chloroplast genomes. Plant Syst Evol. 2008;271:101–22.
Mehmood F, Shahzadi I, Waseem S, Mirza B, Ahmed I, Waheed MT. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics. 2020;112(1):581–91.
Yan M, Zhao X, Zhou J, Huo Y, Ding Y, Yuan Z. The complete chloroplast genomes of Punica granatum and a comparison with other species in Lythraceae. Int J Mol Sci. 2019;20(12):2886.
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo JJ, Zhou S. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 2015;5(1):8348.
Wang J, Fu GF, Tembrock LR, Liao XZ, Ge S, Wu ZQ. Mutational meltdown or controlled chain reaction: The dynamics of rapid plastome evolution in the hyperdiversity of Poaceae. J Syst Evol. 2023;61:328–44.
Yu X, Tan W, Zhang H, Gao H, Wang W, Tian X. Complete chloroplast genomes of Ampelopsis humulifolia and Ampelopsis japonica: molecular structure, comparative analysis, and phylogenetic analysis. Plants. 2019;8(10):410.
Luo Y, Wang R, Li Q, Liu J, Deng Z. The complete chloroplast genome sequence of Nekemias cantoniensis (Hook, Arn.) Planch 1873 (Vitaceae). Mitochondrial DNA B Resour. 2022;7(8):1536–9.
Doyle JJ, Doyle JL. DNA isolation from leaves. Phytochem Bull. 1987;19:11–5.
Lodhi MA, Ye GN, Weeden NF, Reisch BI. A simple and efficient method for DNA extraction from grapevine cultivars and Vitis species. Plant Mol Biol Rep. 1994;12:6–13.
** JJ, Yu WB, Yang JB, Song Y, DePamphilis CW, Yi TS, Li DZ. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:1–31.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W1):W6–11.
Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–74.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57(5):758–71.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–5.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–42.
Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34(17):3030–1.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–302.
Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403.
Zhang Z, Li J, Zhao XQ, Wang J, Wong GKS, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics, proteomics bioinformatics. 2006;4(4):259–63.
Kimura M. The neutral theory of molecular evolution and the world view of the neutralists. Genome. 1989;31(1):24–31.
Fay JC, Wu CI. Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet. 2003;4(1):213–35.
Henriquez CL, Ahmed I, Carlsen MM, Zuluaga A, Croat TB, McKain MR. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics. 2020;112(3):2349–60.
Ruhlman TA, Jansen RK. The plastid genomes of flowering plants. Methods Mol Biol. 2014;1132:3–38.
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK. The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;23(11):2175–90.
Raman G, Park S. The complete chloroplast genome sequence of Ampelopsis: gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front Plant Sci. 2016;7:341.
Cui G, Wang C, Wei X, Wang H, Wang X, Zhu X, Li JH, Yang HS, Duan H. Complete chloroplast genome of Hordeum brevisubulatum: Genome organization, synonymous codon usage, phylogenetic relationships, and comparative structure analysis. PLoS ONE. 2021;16(12):e0261196.
Henriquez CL, Mehmood F, Hayat A, Sammad A, Waseem S, Waheed MT, Maheed MT, Matthews PJ, Croat TB, Poczai P, Ahmed I. Chloroplast genome evolution in the Dracunculus clade (Aroideae, Araceae). Genomics. 2021;113(1):183–92.
Dong WL, Wang RN, Zhang NY, Fan WB, Fang MF, Li ZH. Molecular evolution of chloroplast genomes of orchid species: insights into phylogenetic relationship and adaptive evolution. Int J Mol Sci. 2018;19(3):716.
Ren H, Lu LM, Soejima A, Luke Q, Zhang DX, Chen ZD, Wen J. Phylogenetic analysis of the grape family (Vitaceae) based on the noncoding plastid trnC-petN, trnH-psbA, and trnL-F sequences. Taxon. 2011;60(3):629–37.
Zhang N, Wen J, Zimmer EA. Congruent deep relationships in the grape family (Vitaceae) based on sequences of chloroplast genomes and mitochondrial genes via genome skimming. PLoS ONE. 2015;10(12):e0144701.
Ahmed I, Biggs PJ, Matthews PJ, Collins LJ, Hendy MD, Lockhart PJ. Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol. 2012;4(12):1316–23.
Menezes AP, Resende-Moreira LC, Buzatti RS, Nazareno AG, Carlsen M, Lobo FP, Kalapothakis E, Lovato MB. Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci Rep. 2018;8(1):2210.
Cho WB, Han EK, Choi G, Lee JH. The complete chloroplast genome of Daphne kiusiana, an evergreen broad-leaved shrub on Jeju Island. Conserv Genet Resour. 2018;10:103–6.
Lee SY, Ng WL, Mohamed R, Terhem R. The complete chloroplast genome of Aquilaria malaccensis Lam.(Thymelaeaceae), an important and threatened agarwood-producing tree species. Mitochondrial DNA B Resour. 2018;3(2):1120–1.
Abdullah, Henriquez CL, Mehmood F, Carlsen MM, Islam M, Waheed MT, Pocza P, Croat TB, Ahmed I. Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): unique inverted repeat expansion and contraction affect rate of evolution. J Mol Evol. 2020; 88: 562–574.
Abdullah, Mehmood F, Shahzadi I, Ali Z, Islam M, Naeem M, Mirza B, Lockhart PJ, Ahmed I, Waheed MT. Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J Syst Evol. 2021; 59(2): 388–402.
Lynn A. Gregory and others, molecular basis of diamond-blackfan anemia: structure and function analysis of RPS19. Nucleic Acids Res. 2007;35(17):5913–21.
Aranda-Olmedo I, Tobes R, Manzanera M, Ramos JL, Marqués S. Species-specific repetitive extragenic palindromic (REP) sequences in Pseudomonas putida. Nucleic Acids Res. 2002;30(8):1826–33.
Bachellier S, Perrin D, Hofnung M, Gilson E. Bacterial interspersed mosaic elements (BIMEs) are present in the genome of Klebsiella. Mol Microbiol. 1993;7(4):537–44.
George B, Bhatt BS, Awasthi M, George B, Singh AK. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr Genet. 2015;61:665–77.
Higgins CF, Ames GFL, Barnes WM, Clement JM, Hofnung M. A novel intercistronic regulatory element of prokaryotic operons. Nature. 1982;298(5876):760–2.
Rocco F, De Gregorio E, Di Nocera PP. A giant family of short palindromic sequences in Stenotrophomonas maltophilia. FEMS Microbiol Lett. 2010;308(2):185–92.
Tobes R, Pareja E. Repetitive extragenic palindromic sequences in the Pseudomonas syringae pv. tomato DC3000 genome: extragenic signals for genome reannotation. Res Microbiol. 2005;156(3):424–33.
Tobes R, Ramos JL. REP code: defining bacterial identity in extragenic space. Environ Microbiol. 2005;7(2):225–8.
Peng JY, Zhang XS, Zhang DG, Wang Y, Deng T, Huang XH, Kuang TH, Zhou Q. Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu QE Yang and comparative analyses with other Sinosenecio species. BMC Genomics. 2022;23(1):1–13.
Lu QX, Chang X, Gao J, Wu X, Wu J, Qi ZC, Wang RH, Yan XL, Li P. Evolutionary comparison of the complete chloroplast genomes in Convallaria species and phylogenetic study of asparagaceae. Genes. 2022;13(10):1724.
McDonald MJ, Wang WC, Huang HD, Leu JY. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011;9(6):e1000622.
Xue J, Wang S, Zhou SL. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am J Bot. 2012;99(6):e240–4.
Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11(12):2453–65.
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ. Current trends in microsatellite genoty**. Mol Ecol Resour. 2011;11(4):591–611.
Li J, Tang J, Zeng S, Han F, Yuan J, Yu J. Comparative plastid genomics of four Pilea (Urticaceae) species: insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 2021;21(1):1–13.
Li Y, Dong Y, Liu Y, Yu X, Yang M, Huang Y. Comparative analyses of Euonymus chloroplast genomes: genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front Plant Sci. 2021;11:593984.
Wang HX, Liu H, Moore MJ, Landrein S, Liu B, Zhu ZX, Wang HF. Plastid phylogenomic insights into the evolution of the Caprifoliaceae. Mol Phylogenet Evol. 2020;142:106641.
Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review. Mol Ecol. 2002;11(1):1–16.
Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–99.
**ng ZB, Cao L, Zhou M, **u LS. Analysis on codon usage of chloroplast genome of Eleutherococcus senticosus. Zhongguo Zhong Yao Za Zhi. 2013;38(5):661–5.
Zhou Z, Dang Y, Zhou M. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci. 2016;113(41):E6117–25.
Liu H, He R, Zhang H, Huang Y, Tian M, Zhang J. Analysis of synonymous codon usage in Zea mays. Mol Biol Rep. 2010;37:677–84.
Parvathy ST, Udayasuriyan V, Bhadana V. Codon usage bias. Mol Biol Rep. 2022;49(1):539–65.
Srivastava S, Chanyal S, Dubey A, Tewari AK, Taj G. Patterns of Codon Usage Bias in WRKY Genes of Brassica rapa and Arabidopsis thaliana. J Agric Sci. 2019;11(4):76.
Li X, Wang X, Gong P, Zhang N, Zhang X, Li J. Analysis of codon usage patterns in Giardia duodenalis based on transcriptome data from giardia DB. Genes. 2021;12(8):1169.
Wang P, Mao Y, Su Y, Wang J. Comparative analysis of transcriptomic data shows the effects of multiple evolutionary selection processes on codon usage in Marsupenaeus japonicus and Marsupenaeus pulchricaudatus. BMC Genomics. 2021;22(1):1–14.
Nie X, Deng P, Feng K, Liu P, Du X, You FM, Weining S. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol Biol Report. 2014;32:828–40.
Wang Z, Cai Q, Wang Y, Li M, Wang C, Wang Z, Jiao C, Xu C, Wang H, Zhang Z. Comparative analysis of codon Bias in the chloroplast genomes of Theaceae species. Front Genet. 2022;13:824610.
Asaf S, Khan AL, Khan AR, Waqas M, Kang SM, Khan MA, Lee SM, Lee IJ. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front Plant Sci. 2016;7:843.
Gichira AW, Avoga S, Li Z, Hu G, Wang Q, Chen J. Comparative genomics of 11 complete chloroplast genomes of Senecioneae (Asteraceae) species: DNA barcodes and phylogenetics. Bot Stud. 2019;60(1):1–17.
Wang RN, Milne RI, Du XY, Liu J, Wu ZY. Characteristics and mutational hotspots of plastomes in Debregeasia (Urticaceae). Front Genet. 2020;11:729.
Pascual-Díaz JP, Garcia S, Vitales D. Plastome diversity and phylogenomic relationships in Asteraceae. Plants. 2021;10(12):2699.
Fang J, Lin A, Yuan X, Chen Y, He W, Huang J, Zhang XT, Lin G, Zhang JS, Xue T. The complete chloroplast genome of Isochrysis galbana and comparison with related haptophyte species. Algal Res. 2020;50:101989.
Wu Z, Liao R, Yang T, Dong X, Lan D, Qin R, Liu H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genomics. 2020;21:1–14.
Yang Z, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22(4):1107–18.
Puthiyaveetil S, Ibrahim IM, Jeličić B, Tomašić A, Fulgosi H, Allen JF. Transcriptional control of photosynthesis genes: the evolutionarily conserved regulatory mechanism in plastid genome function. Genome Biol Evol. 2010;2:888–96.
Acknowledgements
The experimental work was conducted at the Smithsonian Laboratories of Analytical Biology of the National Museum of Natural History. The authors acknowledge the Smithsonian High Performance Cluster (SI/HPC; https://doi.org/10.25572/SIHPC) for providing computational resources for the data analyses.
Funding
This study was supported by grants from Natural Sciences Foundation of China (32060055, 31570211) and Natural Sciences Foundation of Hunan Province (2019JJ40232).
Author information
Authors and Affiliations
Contributions
Conceptualization, Lei Zhang and Ze-long Nie; methodology, Lei Zhang; formal analysis, Lei Zhang; investigation, Lei Zhang; resources, Ying Meng; data curation, Da Wang and Guan-Hao He; writing-original draft preparation, Lei Zhang; writing-review and editing, Lei Zhang and Ze-long Nie; visualization, Jun-Ming Zhang; supervision, Ze-long Nie and Jun Wen; project administration, Ying Meng and Ze-long Nie; funding acquisition, Ying Meng. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All plant materials were used following national and international standards and local laws and regulations. The use of all plant materials does not pose any risk to other species in nature. No specific permission is required to collect all samples described in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Meng, Y., Wang, D. et al. Plastid genome data provide new insights into the dynamic evolution of the tribe Ampelopsideae (Vitaceae). BMC Genomics 25, 247 (2024). https://doi.org/10.1186/s12864-024-10149-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10149-w