
Abstract
Background
With more than 36,000 valid fish species, teleost fishes constitute the most species-rich vertebrate clade and exhibit extensive genetic and phenotypic variation, including diverse immune defense strategies. NLRC3 subfamily genes, which are specific to fishes, play vital roles in the immune system of teleosts. The evolution of teleosts has been impacted by several whole-genome duplication (WGD) events, which might be a key reason for the expansions of the NLRC3 subfamily, but detailed knowledge of NLRC3 subfamily evolution in the family Sebastidae is still limited.
Results
Phylogenetic inference of NLRC3 subfamily protein sequences were conducted to evaluate the orthology of NLRC3 subfamily genes in black rockfish (Sebastes schlegilii), 13 other fish species from the families Sebastidae, Serranidae, Gasterosteidae and Cyclopteridae, and three species of high vertebrates (bird, reptile and amphibian). WGD analyses were used to estimate expansions and contractions of the NLRC3 subfamily, and patterns of expression of NLRC3 subfamily genes in black rockfish following bacterial infections were used to investigate the functional roles of these genes in the traditional and mucosal immune system of the Sebastidae. Different patterns of gene expansions and contractions were observed in 17 fish and other species examined, and one and two whole-genome duplication events were observed in two members of family Sebastidae (black rockfish and honeycomb rockfish, Sebastes umbrosus), respectively. Subsequently, 179 copy numbers of NLRC3 genes were found in black rockfish and 166 in honeycomb rockfish. Phylogenetic analyses corroborated the conservation and evolution of NLRC3 orthologues between Sebastidae and other fish species. Finally, differential expression analyses provided evidence of the immune roles of NLRC3 genes in black rockfish during bacterial infections and gene ontology analysis also indicated other functional roles.
Conclusions
We hypothesize that NLRC3 genes have evolved a variety of different functions, in addition to their role in the immune response, as a result of whole genome duplication events during teleost diversification. Importantly, this study had underscored the importance of sampling across taxonomic groups, to better understand the evolutionary patterns of the innate immunity system on which complex immunological novelties arose. Moreover, the results in this study could extend current knowledge of the plasticity of the immune system.
Similar content being viewed by others

Background
Ray-finned teleost fishes occupy the vast majority of the more than 36,000 currently described fish species [1]. Over their 400 million years of evolutionary history [2, 3], teleosts have experienced three rounds of whole-genome duplications (WGD) [4, 5], one more than other vertebrates [6]. Many duplicated genes may lose one of the duplicates via pseudogenization by the accumulation of deleterious mutations during or after duplication events [7,8,9]. Populations of a species that lose different copies of a duplicated gene may become genetically isolated, according to the divergent resolution model of speciation [10, 11]. Duplicated genes might also be retained, rather than lost, and acquire novel functions [12]: this has been suggested as a driving force for major evolutionary transitions, although the evidence for this is mixed [11, 13].
The adaptive immune system is found in all jawed vertebrates [14, 15], but has undergone fundamental modifications of the immune gene repertoire [16,17,18]. Through the loss or doubling of key immune genes during or after WGD events, the immune response system of vertebrate species may have evolved compensatory mechanisms in both adaptive and innate immunity, especially for teleost fishes that contain a number of specific genes because of their unique living environments.
The functional roles of NOD-like receptors (NLRs), key components of the innate and adaptive immune system in invertebrate and vertebrate species, have attracted much research attention. In mammals, there are 20–30 NLR family members [19, 20], while larger numbers of NLR member repertoires have been identified in fish species and other early-diverging metazoans [21, 22]. Based on different types of structural domains, NLRs are usually divided into four subfamilies; NLRA, NLRB, NLRC and NLRP [23]. NLRA subfamilies have been well characterized in teleost fishes including grass carp (Ctenopharyngodon idella) [24], channel catfish (Ictalurus punctatus) [25], and miiuy croaker (Miichthys miiuy) [26]. NLRB and NLRC subfamilies have been also identified preliminarily in several fish species, such as turbot (Scophthalmus maximus L.) [27], miiuy croaker [28] and black rockfish (S. schlegelii) [29]. Meanwhile, the important roles of NLRC genes were also mentioned in several fish species. It is well known that CD4+ T cells were the key component in the immune system, which played as the center in orchestrating adaptive immune responses against pathogenic infections [30, 31]. In Nile tilapia (Oreochromis niloticus), NLRC genes were concentratedly detected in T cells, especially for NLRC3 gene that was mainly observed a high expression level in CD4+ T Cell during LPS/LTA stimulation [32], which might suggest the potential functional roles of NLRC3 gene in teleost adaptive immune response.
According to their physiological functions, NLRs can also be classified into three subgroups; inflammasome-forming, reproductive and regulatory NLRs [33]. Several members of the inflammasome-forming NLRs have been well studied and found to cooperate with the maturation of IL-1β and IL-18 to process pyroptosis [34]. Moreover, regulatory NLRs may perform important functions by acting as either positive or negative regulators on several immune signaling pathways, including the NF-kB and mitogen-activated protein kinase (MAPK) signaling pathway, the type I IFN response and the NOD1-RIPK2 antibacterial pathway [33, 35,36,37]. In fish, regulatory NLRs, including the piscine NLRC3 subfamily proteins, have been also identified and their immune functions investigated [38, 7).
Discussion
While it is known that the small number of NLR proteins in mammals are involved in immune defense and recognize PAMPs [45,46,47], there have been few studies of the larger number of NLR proteins found in taxa other than mammals [28, http://://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/) and conserved domain database (CDD) in the NCBI website [101]. The nucleotide and protein sequences of black rockfish NLRC3 subfamily homologues were extracted from the CDS and protein databases of black rockfish and confirmed by referencing to its genomic database. Python scripts and codes were edited to remove the reduplicated sequences.
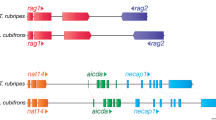
To assess the functions of NLRC3 genes, the immune gene repertoires of the teleost genomes, including several conserved immune genes and several genes interacting with NLRC3 genes, were investigated through a comparative gene mining pipeline comprising BLAST searches, prediction of ORFs and annotation. AP1M2 (adaptor related protein complex 1 subunit mu 2), AP2M1 (adaptor related protein complex 2 subunit mu 1), AP3M2 (adaptor related protein complex 3 subunit mu 2), RIPK2 (receptor interacting serine threonine kinase 2), TLR4 (toll-like receptor 4), TLR9 (toll-like receptor 9), TRAF6 (TNF receptor associated factor 6) and NLRP3 (NACHT, LRR and PYD domains-containing protein 3) were confirmed. Moreover, two highly conserved genes (RAG1: recombination activating gene 1, and RAG2: recombination activating gene 2) were included as controls. Then, the orthologue sequences of these selected genes for nine species, including chicken, Chinese softshell turtle, African clawed frog, lumpfish, yellow perch, nine-spined stickleback, pike, zebrafish and honeycomb rockfish, were collected as queries to determine the copy numbers of these genes in all 17 species using the above methodology.
Phylogenetic analyses of NLRC3 subfamily
To evaluate the phylogenetic orthology of the NLRC3 subfamily, NLRC3 protein sequences of the 14 selected fish species and other high vertebrates (chicken, turtle and frog) were aligned using the MAFFT v.7.505 program. The best-fit substitution model (JTT + F + I + G4) was calculated by IQ-TREE v.2.2.0.3 and a maximum-likelihood phylogenetic tree was constructed on RAxML-NG v.1.1.0. The NLRC3 subfamily sequences of black rockfish were defined and annotated by the phylogenetic relationships between black rockfish and other species and the protein domain architecture on CDD and SMART databases [102]. Two additional phylogenetic analyses were run, using the sequences of NLRC3 genes of black rockfish, honeycomb rockfish and high vertebrates, and only the FISNA domains of the black rockfish to compare the NLRC3 orthology inference. The iTOL online tool was employed to modify the final phylogenetic trees [103].
Functional enrichment of differentially expressed NLRC3 genes
The data for differentially expressed genes of the black rockfish spleen challenged by A. salmonicida, intestine infected by E. tarda and liver with A. salmonicida, at different time points post infection (2 h, 12 and 24 h) were obtained from previous studies [73, 84, 85]. Differential expression analysis of the transcripts was performed using StringTie software and the DESeq R package (3.0.3) [104, 105]. Reads per kilobase of transcript per million mapped reads (RPKM) were obtained. The Benjamini-Hochberg correction procedure was used to adjust the resulting P-value for false discovery rate [106]. The expressed level of transcripts with | log2(Fold Change) | > 0 and P-value < 0.05 were assigned as differentially expressed. Subsequently, based on NLRC3 target sequences of the black rockfish determined above, differential expression patterns for NLRC3 genes were obtained and displayed using Heatmap and volcano plots on the web server of ImageGP [107]. In addition, the transcriptomic data sets of different organs of black rockfish, such as testis and ovary sexual organs in different develo** stages (PRJNA573572), were downloaded and analyzed in the same methods descripted above. The Heatmap plots of NLRC3 genes in these organs were drawn to support the relevant findings in this study.
In order to verify the functional processes in which differentially expressed NLRC3 genes in the black rockfish participate, Gene Ontology (GO) analyses were performed using Blast2GO v.6.0.3 software via a series of analysis schemes [108]. In detail, the query sequences of differentially expressed NLRC3 genes in the three black rockfish transcriptomic databases were matched against the GO annotation database with GO map** after running BLAST at the NCBI. Next, functional annotation was conducted to select GO terms from the GO pool obtained by the map** step and assign them to the query sequences. Then, GO-slim was carried out to functionally summarize a sequence dataset in a uniform and species-specific way. Finally, GO graph visualization was used to generate combined gene ontology annotation graphs in three GO functional categories (Biological Process, Molecular Function, Cellular Component).
Data Availability
The raw data of transcriptome and genome of black rockfish supporting the conclusions of this article were made available by the authors, without undue reservation. The datasets of black rockfish and other species presented in this study can be also found in online repositories (https://www.ncbi.nlm.nih.gov/genome/gdv/). The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The sequences of all NLRC3 genes mentioned in this article were submitted to the GeneBank (https://www.ncbi.nlm.nih.gov/genbank/), and the accession numbers of these NLRC3 sequences are OQ857571 - OQ857749.
References
Eschmeyer’s catalog of fishes. : Genera, species, references. [http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp].
Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, Wainwright PC, Friedman M, Smith WL. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci U S A. 2012;109(34):13698–703.
Betancur RR, Broughton RE, Wiley EO, Carpenter K, López JA, Li C, Holcroft NI, Arcila D, Sanciangco M, Cureton Ii JC et al. The tree of life and a new classification of bony fishes. PLoS Curr 2013, 5.
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, et al. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282(5394):1711–4.
Meyer A, Van de Peer Y. From 2R to 3R: evidence for a fish-specific genome duplication (FSGD). BioEssays. 2005;27(9):937–45.
Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3(10):e314.
Takahata N, Maruyama T. Polymorphism and loss of duplicate gene expression: a theoretical study with application of tetraploid fish. Proc Natl Acad Sci U S A. 1979;76(9):4521–5.
Li WH. Rate of gene silencing at duplicate loci: a theoretical study and interpretation of data from tetraploid fishes. Genetics. 1980;95(1):237–58.
Watterson GA. On the time for gene silencing at duplicate loci. Genetics. 1983;105(3):745–66.
Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5.
Taylor JS, Van de Peer Y, Meyer A. Genome duplication, divergent resolution and speciation. Trends Genet. 2001;17(6):299–301.
Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151(4):1531–45.
Magadum S, Banerjee U, Murugan P, Gangapur D, Ravikesavan R. Gene duplication as a major force in evolution. J Genet. 2013;92(1):155–61.
Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet. 2010;11(1):47–59.
Dornburg A, Yoder JA. On the relationship between extant innate immune receptors and the evolutionary origins of jawed vertebrate adaptive immunity. Immunogenetics. 2022;74(1):111–28.
Star B, Nederbragt AJ, Jentoft S, Grimholt U, Malmstrøm M, Gregers TF, Rounge TB, Paulsen J, Solbakken MH, Sharma A, et al. The genome sequence of Atlantic Cod reveals a unique immune system. Nature. 2011;477(7363):207–10.
Buonocore F, Gerdol M. Alternative adaptive immunity strategies: coelacanth, cod and shark immunity. Mol Immunol. 2016;69:157–69.
Malmstrøm M, Matschiner M, Tørresen OK, Star B, Snipen LG, Hansen TF, Baalsrud HT, Nederbragt AJ, Hanel R, Salzburger W, et al. Evolution of the immune system influences speciation rates in teleost fishes. Nat Genet. 2016;48(10):1204–10.
Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285–7.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32.
Hibino T, Loza-Coll M, Messier C, Majeske AJ, Cohen AH, Terwilliger DP, Buckley KM, Brockton V, Nair SV, Berney K, et al. The immune gene repertoire encoded in the purple sea urchin genome. Dev Biol. 2006;300(1):349–65.
Howe K, Schiffer PH, Zielinski J, Wiehe T, Laird GK, Marioni JC, Soylemez O, Kondrashov F, Leptin M. Structure and evolutionary history of a large family of NLR proteins in the zebrafish. Open Biol. 2016;6(4):160009.
Williams A, Flavell RA, Eisenbarth SC. The role of NOD-like receptors in sha** adaptive immunity. Curr Opin Immunol. 2010;22(1):34–40.
Chen WQ, Xu QQ, Chang MX, Nie P, Peng KM. Molecular characterization and expression analysis of nuclear oligomerization domain proteins NOD1 and NOD2 in grass carp Ctenopharyngodon idella. Fish Shellfish Immunol. 2010;28(1):18–29.
Rajendran KV, Zhang J, Liu S, Kucuktas H, Wang X, Liu H, Sha Z, Terhune J, Peatman E, Liu Z. Pathogen recognition receptors in channel catfish: I. Identification, phylogeny and expression of NOD-like receptors. Dev Comp Immunol. 2012;37(1):77–86.
Li J, Kong L, Gao Y, Wu C, Xu T. Characterization of NLR-A subfamily members in miiuy croaker and comparative genomics revealed NLRX1 underwent duplication and lose in Actinopterygii. Fish Shellfish Immunol. 2015;47(1):397–406.
Zhang L, Cao M, Li Q, Yan X, Xue T, Song L, Su B, Li C. Genome-wide identification of NOD-like receptors and their expression profiling in mucosal tissues of turbot (Scophthalmus maximus L.) upon bacteria challenge. Mol Immunol. 2021;134:48–61.
Li J, Chu Q, Xu T. A genome-wide survey of expansive NLR-C subfamily in miiuy croaker and characterization of the NLR-B30.2 genes. Dev Comp Immunol. 2016;61:116–25.
Cao M, Yan X, Li Q, Fu Q, Yang N, Song L, Li C. Genome-wide identification and analysis of NOD-like receptors and their potential roles in response to Edwardsiella tarda Infection in black rockfish (Sebastes schlegelii). Aquaculture. 2021;541:736803.
Zhu X, Zhu J. CD4 T helper cell subsets and related human immunological disorders. Int J Mol Sci 2020, 21(21).
Yang W, Chen X, Hu H. CD4(+) T-cell differentiation in vitro. Methods Mol Biol. 2020;2111:91–9.
Li Q, Jiang B, Zhang Z, Huang Y, Xu Z, Chen X, Huang Y, Jian J, Yan Q. Involvement and characterization of NLRCs and pyroptosis-related genes in Nile tilapia (Oreochromis niloticus) immune response. Fish Shellfish Immunol. 2022;130:602–11.
Coutermarsh-Ott S, Eden K, Allen IC. Beyond the inflammasome: regulatory NOD-like receptor modulation of the host immune response following virus exposure. J Gen Virol. 2016;97(4):825–38.
Davis BK, Philipson C, Hontecillas R, Eden K, Bassaganya-Riera J, Allen IC. Emerging significance of NLRs in inflammatory bowel Disease. Inflamm Bowel Dis. 2014;20(12):2412–32.
Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, et al. NLRX1 protein attenuates inflammatory responses to Infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity. 2011;34(6):854–65.
Tocker AM, Durocher E, Jacob KD, Trieschman KE, Talento SM, Rechnitzer AA, Roberts DM, Davis BK. The scaffolding protein IQGAP1 interacts with NLRC3 and inhibits type I IFN production. J Immunol. 2017;199(8):2896–909.
Fang H, Wu XM, Hu YW, Song YJ, Zhang J, Chang MX. NLRC3-like 1 inhibits NOD1-RIPK2 pathway via targeting RIPK2. Dev Comp Immunol. 2020;112:103769.
Song L, Gao C, Xue T, Yang N, Fu Q, Zhu Q, Ge X, Li C. Characterization and expression analysis of mitochondrial localization molecule: NOD-like receptor X1 (Nlrx1) in mucosal tissues of turbot (Scophthalmus maximus) following bacterial challenge. Dev Comp Immunol. 2021;116:103944.
Chang MX, **ong F, Wu XM, Hu YW. The expanding and function of NLRC3 or NLRC3-like in teleost fish: recent advances and novel insights. Dev Comp Immunol. 2021;114:103859.
Álvarez CA, Ramírez-Cepeda F, Santana P, Torres E, Cortés J, Guzmán F, Schmitt P, Mercado L. Insights into the diversity of NOD-like receptors: identification and expression analysis of NLRC3, NLRC5 and NLRX1 in rainbow trout. Mol Immunol. 2017;87:102–13.
Liu J, Lu L, Liu L, Chen D, Yang F, Geng Y, Li Z, Huang X, Ouyang P, Wang J, et al. Genomic structure and molecular characterization of NLRC3-like from Siberian sturgeon (Acipenser baerii) and expression response to Streptococcus iniae and pathogen-associated molecular patterns. Fish Shellfish Immunol Rep. 2021;2:100042.
Zhou F, Zhan Q, Ding Z, Su L, Fan J, Cui L, Chen N, Wang W, Liu H. A NLRC3-like gene from blunt snout bream (Megalobrama amblycephala): molecular characterization, expression and association with resistance to Aeromonas hydrophila Infection. Fish Shellfish Immunol. 2017;63:213–9.
Sun J, Zhao X, Pei C, Zhu L, Zhang J, Kong X. Molecular characterization of NLRC3 and its interaction with other inflammasome components and regulation on the bacterial colonization in Qihe crucian carp Carassius auratus. Fish Shellfish Immunol. 2022;131:958–71.
Zhang L, Gao Z, Yu L, Zhang B, Wang J, Zhou J. Nucleotide-binding and oligomerization domain (NOD)-like receptors in teleost fish: current knowledge and future perspectives. J Fish Dis. 2018;41(9):1317–30.
Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 2003;5(9):581–92.
Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442(7098):39–44.
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and Disease. Nat Immunol. 2006;7(12):1250–7.
Hui F, Guo S, Liu J, Li M, Geng M, **a Y, Liu X, Li Q, Li J, Zhu T. Genome-wide identification and characterization of NLR genes in lamprey (Lethenteron reissneri) and their responses to lipopolysaccharide/poly(I:C) challenge. Mol Immunol. 2022;143:122–34.
Blomme T, Vandepoele K, De Bodt S, Simillion C, Maere S, Van de Peer Y. The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006;7(5):1–12.
Cañestro C, Albalat R, Irimia M, Garcia-Fernàndez J. Impact of gene gains, losses and duplication modes on the origin and diversification of vertebrates. In: Semin Cell Dev Biol: 2013: Elsevier; 2013: 83–94.
Glasauer SM, Neuhauss SC. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol Genet Genomics. 2014;289(6):1045–60.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu Rubripes. Science. 2002;297(5585):1301–10.
Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431(7011):946–57.
Schartl M, Walter RB, Shen Y, Garcia T, Catchen J, Amores A, Braasch I, Chalopin D, Volff JN, Lesch KP. The genome of the platyfish, **phophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet. 2013;45(5):567–72.
Van Gorp H, Kuchmiy A, Van Hauwermeiren F, Lamkanfi M. NOD-like receptors interfacing the immune and reproductive systems. FEBS J. 2014;281(20):4568–82.
Slim R, Coullin P, Diatta AL, Chebaro W, Courtin D, Abdelhak S, Garcia A. NLRP7 and the genetics of post-molar choriocarcinomas in Senegal. Mol Hum Reprod. 2012;18(1):52–6.
Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P. Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Advances in bioinformatics 2008, 2008.
Sena P, Riccio M, Marzona L, Nicoli A, Marsella T, Marmiroli S, Bertacchini J, Fano RA, La Sala GB, De Pol A. Human MATER localization in specific cell domains of oocytes and follicular cells. Reprod Biomed Online. 2009;18(2):226–34.
Wang C, Dixon P, Decordova S, Hodges M, Sebire N, Ozalp S, Fallahian M, Sensi A, Ashrafi F, Repiska V. Identification of 13 novel NLRP7 mutations in 20 families with recurrent hydatidiform mole; missense mutations cluster in the leucine-rich region. J Med Genet. 2009;46(8):569–75.
Peng H, Chang B, Lu C, Su J, Wu Y, Lv P, Wang Y, Liu J, Zhang B, Quan F. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS ONE. 2012;7(1):e30344.
Laing KJ, Purcell MK, Winton JR, Hansen JD. A genomic view of the NOD-like receptor family in teleost fish: identification of a novel NLR subfamily in zebrafish. BMC Evol Biol. 2008;8:42.
Li X, Gold B, O’HUigin C, Diaz-Griffero F, Song B, Si Z, Li Y, Yuan W, Stremlau M, Mische C, et al. Unique features of TRIM5alpha among closely related human TRIM family members. Virology. 2007;360(2):419–33.
Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. The B30.2 domain of pyrin, the familial Mediterranean Fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103(26):9982–7.
Song B, Gold B, O’Huigin C, Javanbakht H, Li X, Stremlau M, Winkler C, Dean M, Sodroski J. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J Virol. 2005;79(10):6111–21.
Loeven NA, Medici NP, Bliska JB. The pyrin inflammasome in host-microbe interactions. Curr Opin Microbiol. 2020;54:77–86.
Conti BJ, Davis BK, Zhang J, O’Connor W Jr., Williams KL, Ting JP. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J Biol Chem. 2005;280(18):18375–85.
Gültekin Y, Eren E, Özören N. Overexpressed NLRC3 acts as an anti-inflammatory cytosolic protein. J Innate Immun. 2015;7(1):25–36.
Eren E, Berber M, Özören N. NLRC3 protein inhibits inflammation by disrupting NALP3 inflammasome assembly via competition with the adaptor protein ASC for pro-caspase-1 binding. J Biol Chem. 2017;292(30):12691–701.
Wu S, Huang J, Li Y, Liu Z, Zhao L. Integrated analysis of lncRNA and circRNA mediated ceRNA regulatory networks in skin reveals innate immunity differences between wild-type and yellow mutant rainbow trout (Oncorhynchus mykiss). Front Immunol. 2022;13:802731.
Paria A, Deepika A, Sreedharan K, Makesh M, Chaudhari A, Purushothaman CS, Thirunavukkarasu AR, Rajendran KV. Identification of nod like receptor C3 (NLRC3) in Asian seabass, lates calcarifer: Characterisation, ontogeny and expression analysis after experimental Infection and ligand stimulation. Fish Shellfish Immunol. 2016;55:602–12.
Hu S, Du X, Huang Y, Fu Y, Yang Y, Zhan X, He W, Wen Q, Zhou X, Zhou C, et al. NLRC3 negatively regulates CD4 + T cells and impacts protective immunity during Mycobacterium tuberculosis Infection. PLoS Pathog. 2018;14(8):e1007266.
Krishnan R, Rajendran R, Jang YS, Kim JO, Yoon SY, Oh MJ. NLRC3 attenuates antiviral immunity and activates inflammasome responses in primary grouper brain cells following nervous necrosis virus Infection. Fish Shellfish Immunol. 2022;127:219–27.
Gao C, Cai X, Ma L, Li C. Identification of mRNA-miRNA-lncRNA regulatory network associated with the immune response to Aeromonas salmonicides Infection in the black rockfish (Sebastes schlegelii). Dev Comp Immunol. 2022;130:104357.
Cheng D, Xu JH, Li JY, Wang SY, Wu TF, Chen QK, Yu T. Butyrate ameliorated-NLRC3 protects the intestinal barrier in a GPR43-dependent manner. Exp Cell Res. 2018;368(1):101–10.
Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, Sharma BR, Qi X, Pelletier S, Vogel P, et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature. 2016;540(7634):583–7.
Karki R, Malireddi RKS, Zhu Q, Kanneganti TD. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of Colorectal cancer. Cell Cycle. 2017;16(13):1243–51.
Li M, Wang Q-l, Lu Y, Chen S-l, Li Q, Sha Z-x. Expression profiles of NODs in channel catfish (Ictalurus punctatus) after Infection with Edwardsiella tarda, Aeromonas hydrophila, Streptococcus iniae and channel catfish Hemorrhage reovirus. Fish Shellfish Immunol. 2012;33(4):1033–41.
Hou Z, Ye Z, Zhang D, Gao C, Su B, Song L, Tan F, Song H, Wang Y, Li C. Characterization and expression profiling of NOD-like receptor C3 (NLRC3) in mucosal tissues of turbot (Scophthalmus maximus L.) following bacterial challenge. Fish Shellfish Immunol. 2017;66:231–9.
Li ZT, Liu H, Zhang WQ. NLRC3 alleviates hypoxia/reoxygenation induced inflammation in RAW264.7 cells by inhibiting K63-linked ubiquitination of TRAF6. Hepatobiliary Pancreat Dis Int. 2020;19(5):455–60.
Ma YY, Zhang GH, Li J, Wang SB, Hu ZM, Zhang CW, Li E. The correlation of NLRC3 expression with the progression and prognosis of hepatocellular carcinoma. Hum Pathol. 2018;82:273–81.
Kang JH, Li MJ, Luan PP, Jiang DK, Chen YW, Xu X, Yu Q, Xu YW, Su Q, Peng WH, et al. NLRC3 silencing accelerates the invasion of hepatocellular carcinoma cell via IL-6/JAK2/STAT3 pathway activation. Cell Biol Int. 2020;44(10):2053–64.
Zhang M, Cao M, **u Y, Fu Q, Yang N, Su B, Li C. Identification of antimicrobial peptide genes in black rockfish Sebastes schlegelii and their responsive mechanisms to Edwardsiella tarda Infection. Biology (Basel) 2021, 10(10).
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):119.
Cao M, Yan X, Su B, Yang N, Fu Q, Xue T, Song L, Li Q, Li C. Integrated analysis of circRNA-miRNA-mRNA regulatory networks in the intestine of Sebastes schlegelii following Edwardsiella tarda challenge. Front Immunol. 2020;11:618687.
Zhang X, Cao M, Xue T, Yu H, Yang T, Yan X, Li C. Characterization of IL-17/IL-17R gene family in Sebastes schlegelii and their expression profiles under Aeromonas salmonicida and Edwardsiella piscicida Infections. Aquaculture. 2022;551:737901.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16(1):157.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238.
Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34(14):2490–2.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74.
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35(21):4453–5.
Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. 2010;17(3):337–54.
Lemoine F, Domelevo Entfellner JB, Wilkinson E, Correia D, Dávila Felipe M, De Oliveira T, Gascuel O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature. 2018;556(7702):452–6.
FishBase. [https://www.fishbase.org.au/v4].
Gao F, Chen C, Arab DA, Du Z, He Y, Ho SYW. EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol Evol. 2019;9(7):3891–8.
Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34(7):1812–9.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Mendes FK, Vanderpool D, Fulton B, Hahn MW. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020.
Sun P, Jiao B, Yang Y, Shan L, Li T, Li X, ** Z, Wang X, Liu J. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. bioRxiv 2021:2021.2004.2029.441969.
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48(D1):D265–d268.
Letunic I, Khedkar S, Bork P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2020;49(D1):D458–60.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300.
Chen T, Liu Y, Huang L. ImageGP: an easy-to-use data visualization web server for scientific researchers. iMeta. 2022;1(1):e5.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Acknowledgements
We thank the Fish Immunology Laboratory of Qingdao Agricultural University for providing the genome and protein sequence data of black rockfish.
Funding
This study was supported by National Natural Science Foundation of China (No. 32073005), Young Experts of Taishan Scholars (NO. tsqn 201909130), Science and Technology Support Plan for Youth Innovation of Colleges and Universities in Shandong Province (2019KJF003), and was also supported by Shandong Technical System of Fish Industry (SDAIT-12-03).
Author information
Authors and Affiliations
Contributions
Chengbin Gao: Formal analysis, Methodology, Conceptualization, Data Curation, Writing - Original Draft. **n Cai: Formal analysis, Methodology, Software, Validation, Data Curation. Alan J. Lymbery: Software, Validation, Writing - Review & Editing. Le Ma: Validation, Investigation. Chao Li: Conceptualization, Visualization, Supervision, Project administration, Funding acquisition.All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols involving the use of animals were reviewed and approved by the Murdoch University Animal Ethics Committee (Permit No. N3359/21). All the methods used in this study were carried out in accordance with approved protocol.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, C., Cai, X., Lymbery, A.J. et al. The evolution of NLRC3 subfamily genes in Sebastidae teleost fishes. BMC Genomics 24, 683 (2023). https://doi.org/10.1186/s12864-023-09785-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09785-5