
Abstract
Background
Monochasma savatieri Franch. ex Maxim is a medicinally valuable herb. However, the collection and protection of the wild germplasm resources of M. savatieri are still insufficient, and their genetic diversity and population structure have been poorly studied.
Results
We collected and examined 46 M. savatieri individuals from Fujian, Hunan, Jiangxi, and Zhejiang provinces for genetic diversity and population structure, using 33 newly developed expressed sequence tag-simple sequence repeat (EST-SSR) markers. Applying these markers, we detected a total of 208 alleles, with an average of 6.303 alleles per locus. The polymorphic information content varied from 0.138 to 0.884 (average: 0.668), indicating a high level of polymorphism. At the population level, there was a low degree of genetic diversity among populations (I = 0.535, He = 0.342), with Zhejiang individuals showing the highest genetic diversity among the four populations (Fst = 0.497), which indicated little gene flow within the M. savatieri populations (Nm = 0.253). Mantel test analysis revealed a significant positive correlation between geographical and genetic distance among populations (R2 = 0.3304, p < 0.05), and structure and principal coordinate analyses supported classification of populations into three clusters, which was consistent with the findings of cluster analysis.
Conclusions
As a rare medicinal plants, the protection of M. savatieri does not look optimistic, and accordingly, protective efforts should be beefed up on the natural wild populations. This study provided novel tools and insights for designing effective collection and conservation strategies for M. savatieri.
Similar content being viewed by others

Background
Monochasma savatieri Franch. ex Maxim, a perennial medicinal plant in the Scrophulariaceae family, is endemic to China and Japan, where it grows preferentially on sandy hillsides and in grassy clusters. In China, it is mainly distributed in southeastern regions, including the provinces of Jiangxi, Fujian, Hunan, and Zhejiang [1, 2]. The whole plant of M. savatieri can be used for medicinal purposes, including the treatment of colds, cough, pneumonia, fever, toothache, and irregular menstruation [3,4,5]. Preparations of the plant are characterized by a wide range of biological activities, including antioxidant, antibacterial, and antiviral activities, which are assumed to be attributed the main medicinally effective components, namely, phenylpropanoids, flavonoids, alkaloids, saponins, and polysaccharides [6, 7].
M. savatieri has a long history of medicinal use and is one of the essential component of Yanning granules, which has been designated a “national protected traditional Chinese medicine variety” [8, 9]. However, as an important and valuable medicinal plant, the artificial cultivation of M. savatieri has been unsuccessful. In recent years, there has been an increase in the market demand for M. savatieri, which has often led to limitations in the supply of its wild resources. Such scarcities among wild populations can be attributed to the fact that M. savatieri is a root hemiparasite that has stringent habitat requirements and a naturally weak reproductive capacity [10]. Moreover, the seeds M. savatieri are characterized by a short period of dormancy, and germination rates decline sharply with a prolongation of storage time. Consequently, if, on reaching maturity, the seeds lack suitable germination conditions, they will rapidly lose vitality [11]. Owing to a combination of unfavorable factors, including over-harvesting and habitat destruction, the natural habitats of wild resources of M. savatieri have sharply deteriorated and reduced in size. Their number of individuals and size of natural population have markedly declined in recent years owing to the short seed dormancy and inherently poor regeneration. This has resulted in substantial losses of genetic resources, as indicated by a survey conducted in China by Chen et al. [12], who found that wild resources of M. savatieri in almost all investigated regions showed sharp declines. Moreover, in Japan, M. savatieri has long been listed as an endangered species [13]. Consequently, conservation of the wild resources of M. savatieri is considered a matter of particular urgency. Currently, however, there is a notable lack of information regarding the genetic variation of M. savatieri, which thereby hinders evaluations of the genetic status of this species and makes it difficult to formulate appropriate scientifically based resource conservation strategies.
Analyses of species genetic diversity and population structure play key roles in genetic resource conservation and plant breeding [14, 15]. In this regard, a range of molecular markers, including random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), sequence-related amplified polymorphism (SRAP), simple sequence repeat (SSR), and single-nucleotide polymorphism (SNP) markers, have been widely used to assess the genetic diversity of species and reveal their population structures [16,17,28,29,30]. To date, studies on the genetic diversity of M. savatieri are still limited, and one of the most important reasons is the lack of effective molecular markers for the species.
In this study, great efforts have been made to collect and preserve the wild germplasm resources of M. savatieri from the major distribution provinces of China. Meanwhile, we detected an abundance of EST-SSRs in the assembled transcripts of the full-length transcriptome data of M. savatieri, and from among these, we developed reproducible polymorphic EST-SSR markers to assess the level of genetic diversity and genetic structure for the collected wild germplasm resources of M. savatieri. The collected wild germplasm resources and genetic information will provide novel insights for the conservation, utilization and breeding strategies development of M. savatieri.
Results
Characterization of EST-SSR loci
In total, we detected 35,807 EST-SSR loci distributed among 18,279 of a total of 40,970 transcript sequences (98.98 Mb) (The data were deposited and are available at https://github.com/AWan222/LRCyc). The frequency of SSR distribution (number of SSR/total transcripts) was 87.40%, with an average of one locus per 2.76 kb. In addition, we found that 9078 sequences contained more than one SSR loci, and that there were 6363 (17.77%) compound SSRs. The SSR types were abundant, ranging from mononucleotide to hexanucleotide repeats, and the number of each type varied. Among these, mononucleotide (18,578, 51.88%) and dinucleotide (12,750, 35.61%) EST-SSRs were the most abundant repeat types, followed by trinucleotide loci (4171, 11.65%), with less than 1.00% of the total being represented by tetra-, penta-, and hexa-nucleotides (Table 1). The number of repeats varied greatly among the different repeat types of SSRs. The highest frequency of SSR repeat types occurred with 10 repeats, with 6466, accounting for 18.06%, followed by 6 (4504, 12.58%) and 11 (3797, 10.60%) repeats (Fig. 1). The repeats of mononucleotide SSRs were mainly concentrated in the range of 10–20 times, and the number of other SSR repeats were mainly concentrated in the range of 5–10 times. Among the identified SSRs, a total of 95 types of motif were detected (Fig. 1). Tetra- and hexa-nucleotide repeat types exhibited the highest number of motif types (21 and 39 types, respectively). Moreover, among different repeat motif types, the A/T repeat motif was the most abundant (17,737, 49.54%) followed by AG/CT (7445, 20.79%), AC/GT (2867, 8.01%), AT/AT (2406, 6.72%), and AAT/ATT (951, 2.66%). These findings provide a solid foundation for the development of EST-SSR markers in M. savatieri.

Characterization of SSR loci. A Distribution of SSR repeat number in Monochasma savatieri. B Distribution of SSR motif types in Monochasma savatieri
Genetic diversity
For the purposes of EST-SSR primers screening, we randomly selected the DNA of 10 different M. savatieri individuals, among which, we developed 33 highly polymorphic SSRs that were subsequently used to assess the genetic diversity and genetic relationships among the 46 M. savatieri specimens obtained from different wild habitats in China (Table 2). We accordingly succeeded in amplifying a total of 208 alleles, with the numbers of alleles scored for the 33 loci ranging from 3 (LRC-60747-1) to 11 (LRC-19013-3) with an average of 6.303 (Fig. 2). The average MAF ranged from 0.130 to 0.924 (average: 0.424), whereas the GD ranged from 0.143 to 0.894 (average: 0.706), thereby indicating differences in the polymorphisms of the EST-SSR loci. The PIC ranged from 0.138 to 0.884 (average: 0.668), with the values indicating that 31 SSR loci (93.94%) were highly polymorphic (PIC ≥0.5). Of the remaining two loci, one was moderately polymorphic (0.25 < PIC < 0.5) and the other showed low polymorphism (PIC ≤0.25).

Results of the PCR amplification of 46 M. Monochasma savatieri germplasm resources using the EST-SSR markers (A) LRC-19013-3, (B) LRC-35320-1, and (C) LRC-10353-1. M: DNA Ladder; 1 to 46: materials of the 46 Monochasma savatieri individuals listed in Supplemental Table 1
The results obtained for population-level indexes of genetic diversity in each population are shown in Table 3. The number of alleles (Na) for each locus at the population level ranged from 1.909 to 2.273 (average: 2.068), whereas the number of effective alleles (Ne) ranged from 1.682 to 1.823 (mean: 1.747). Among the four populations, we recorded the highest values of Na and Ne in the ZJ and JX populations, respectively. Expected heterozygosity (He) at the population level varied from 0.314 to 0.362 (mean: 0.342) and Shannon index (I) values obtained for the four populations ranged from 0.481 to 0.583 (mean 0.535), with both indexes indicating the highest genetic diversity in the ZJ population, whereas the FJ population was characterized by the lowest genetic diversity. Furthermore, analysis of the percentage of polymorphic loci (PPL) revealed that the FJ and ZJ populations had the lowest (63.64%) and highest (75.76%) PPL values respectively, with a mean value among populations of 70.45%. These results accordingly indicated that the ZJ population has the highest genetic diversity, whereas comparatively, the JX, HN, and FJ populations are characterized by relatively lower genetic diversities.
Sample size is an important factor in the analysis of genetic diversity, because it affects genetic diversity indexes. To verify the accuracy of the genetic diversity results using 46 M. savatieri samples, we randomly selected different individuals from 46 samples and constructed 26 groups of different sample sizes with three replicates for each group to plot the trends of the Ne, I and He indexes. As shown in Fig. 3, the genetic diversity indexes Ne, I and He increased with an increase in the effective sample size. However, when the sample size reached approximately 25, the genetic diversity indexes stabilized and no longer increased significantly with increase in sample size. Therefore, the genetic diversity analysis with the 46 M. savatieri samples in this study was accurate and effective.

Fitting curves between sample size and different genetic diversity indexes
Genetic differentiation
To gain estimates of genetic differentiation, we performed AMOVA analysis at three levels, namely, among populations, within populations, and within individuals. We accordingly established that 49.74% of the total genetic variation originate from variability within the populations, whereas 48.36% is attributed to differences among populations, and only 1.91% ascribed to within-individual differences (Table 4). F-statistics analysis revealed that the inbreeding coefficient (Fis) and overall fixation index (Fit) were 0.962 and 0.981, respectively, and that the genetic differentiation coefficient (Fst) was 0.497 (Fst > 0.25), indicating a relatively large genetic differentiation among the populations. In addition, we determined a gene flow (Nm) value of 0.253 (Nm < 1), which tends to indicate that the genetic differentiation among population may be caused by migration or genetic drift.
Population structure
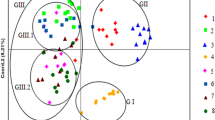
To gain an understanding of the structural and genetic characteristics of the M. savatieri populations, we subjected the 46 target individuals to PCoA, UPGMA, and structure analyses. Our PCoA analysis was based on the use of a correlation genetic similarity matrix, showing the first three principal coordinates with Eigen value effects of 20.77, 18.23, and 11.78% respectively, the derived scatter plot of which discriminately divided the 46 individuals into three main groups (Fig. 4). Notably, these three PcoA clusters appeared to be identical to those identified based on structure analysis, representing the natural distribution of M. savatieri. Structure clustering analysis revealed that the maximal ΔK value (120.776) was obtained for a K value of 3 (Fig. 5), thereby indicating that the 46 M. savatieri individuals could be separated into three distinct genetic clusters (Fig. 5). Specifically, the genetic structure map revealed that the nine individuals in the FJ population grouped in Cluster 1 (blue cluster), the 19 individuals in the HN and JX populations grouped in Cluster 2 (green cluster), and the remaining 18 individuals from the ZJ population were collected in Cluster 3 (red cluster). Consistent with the results of the PCoA analysis, these findings thus indicated that gene penetration among the different M. savatieri populations was relatively low.

A principal coordinate analysis (PCoA) plot for the four Monochasma savatieri populations showing the separation into three main clusters

A The structure of the Monochasma savatieri population inferred based on Bayesian clustering of the EST-SSR markers data. B A graph showing the relationship between ΔK and K values. C The relationship between the pairwise estimates of genetic distance and the corresponding geographical distances among the four populations of Monochasma savatieri
The same three-way partitioning of populations was similarly confirmed by the findings of UPGMA cluster analysis. The UPGMA dendrogram was constructed based on Nei’s genetic distance and accurately reflects the genetic relationships among and within populations. The tree showed that the 46 M. savatieri individuals from the four populations could be divided into two major clusters (Fig. 6), with Cluster 1 comprising exclusively the FJ population and Cluster 2 containing all individuals from the remaining three populations, which were further divided into two short branches, with populations (HN and JX) forming one short branch and the ZJ population comprising the other. Furthermore, both the UPGMA dendrogram and STRUCTURE clustering indicated that the clustering of populations tended to be consistent with the geographical distance among populations. These observations were supported by our Mantel test results, which indicated a significant correlation between genetic and geographical distances (R2 = 0.3304, p < 0.05) (Fig. 5).

A UPGMA dendrogram of the 46 Monochasma savatieri individuals
Discussion
M. savatieri is an important and rare medicinal plant found only in southeast China and the islands of Kyushu in southwestern Japan [12]. In recent years, as the market demand for M. savatieri increased, overharvesting and habitat destruction caused its wild germplasm resources to reduce excessively. Therefore, it is of great importance to collect and analyze the germplasm resources of M. savatieri for the formulation of protection strategies and utilization of the wild resources. In this study, great efforts were made to collect and preserve the wild germplasm resources of M. savatieri from the major distribution provinces of China. This is the most reported current collection for the wild germplasm resources of M. savatieri. The abundant germplasm resources of M. savatieri will provide considerable opportunities for future genetic research and breeding application.
Molecular markers play a pivotal role in genetic research and molecular breeding. However, available resources for molecular markers in M. savatieri are very scarce. In this study, we developed a novel set of EST-SSR markers and used them to determine the genetic diversity of M. savatieri populations. To the best of our knowledge, this is the first study that has sought to assess the genetic diversity of this important medicinal plant species. The frequency of EST-SSRs in M. savatieri is comparable to that of found in Rhododendron fortunei [31] and Cocos nucifera [32], although somewhat lower than that previously reported for Pongamia pinnata [33] and Stephanandra incisa [34], but notably higher than that in Paeonia suffruticosa [35] and Cephalotaxus oliveri [36]. These findings accordingly reveal the relatively broad range of SSR densities among plants at the species level. With respect to M. savatieri, we identified mono- and dinucleotides as the predominant types of EST-SSR repeat. This pattern contrasts to a certain extent with that in a range of other plants, including Corchorus spp. [37], Lycium barbarum [38], and Curcuma alismatifolia [39], for which di- and tri-nucleotide repeats predominate. We speculate that this disparity could be attributable to the relatively long evolutionary history of M. savatieri [40]. Furthermore, among the different repeat motifs, we identified A/T (17,737, 49.54%) as the predominant type in M. savatieri. Notably, the proportion of A/T motifs was found to be considerably higher than that of G/C motifs, which is consistent with the pattern reported for numerous plants, such as Opisthopappus [41], Pinus koraiensis [42], and Phoebe bournei [43].
Genetic diversity is an important prerequisite for species adaptation to environmental change and the development of disease resistance, the levels of which determine the viability and evolutionary potential of populations, and are often used to predict species trends and potential dangers [44]. The use of molecular markers, such as RAPD, AFLP, and SSR, has been shown to be an accurate and reliable approach for analyzing the genetic diversity of plant populations [45], among which, SSRs are widely employed owing to their strengths. In this study, we used 33 novel developed EST-SSR primers to examine the genetic diversity of M. savatieri specimens collected from 10 geographically separated wild areas, the results of which indicated a large variation in the number of alleles of each locus, ranging from 3 to 11 with a mean value of 6.303. The average GD and PIC values determined for the loci, 0.706 and 0.668, respectively, were found to be higher than those reported for some plants, such as those in the genera Polygonatum [46] and Perilla [47], indicating the high polymorphism of these loci. Additionally, the differences in the values for each locus indicate that these 33 loci are characterized by significant genetic variation among the populations, thereby highlighting their importance with respect to the evaluation of M. savatieri germplasm resources. The ploidy level, defined as the number of sets of chromosomes in the nucleus, is a crucial characteristics for studying biodiversity and develo** strategies for plant breeding. In this study, within the expected PCR product sizes, the overwhelming majority of alleles of amplification loci of the 33 SSR markers in each germplasm were one or two, whereas three alleles existed only in a few loci. Therefore, it was speculated that M. savatieri was a diploid or triploid plant, but additional approaches, including flow cytometry and chromosome counting, were requested to further define the ploidy level of M. savatieri.
Analysis of M. savatieri genetic diversity at the population level revealed that He and I ranged from 0.314 to 0.362 (average: 0.342) and 0.481 to 0.583 (average: 0.535) respectively, which are lower than the values previously obtained for Paeonia decomposita [48], Magnolia sinostellata [49], and Camellia nitidissima [50], thus indicating that the genetic diversity of M. savatieri is lower than that of these three species. For plants, reproductive patterns, genetic drift, natural selection, anthropogenic activities, and habitat fragmentation are among the main factors contributing to changes in genetic diversity [51]. It can be speculated that the low genetic diversity of M. savatieri populations is attributable to its narrow range of distribution, along with the large spatial distance between populations, which limits inter-population gene exchange. Moreover, excessive harvesting of natural populations, habitat destruction, and declining population sizes may contribute to inbreeding and genetic drift within the M. savatieri population, thereby reducing its genetic diversity. Nevertheless, among the four regional populations examined in this study, we identified the Zhejiang population as being the genetically most diverse. Generally, a higher genetic diversity is indicative of a higher complexity of plant diversity, and consequently a greater potential for environmental adaptation [44]. Accordingly, analysis of the genetic diversity of M. savatieri will predictably provide a theoretical basis for its protection and breeding.
The degree of differentiation between natural populations can be described in terms of gene flow (Nm) and the genetic differentiation coefficient (Fst), which are negatively correlated, with a higher differentiation coefficient between populations being taken to be indicative of a lower extent of gene flow [52]. Genetic differentiation among plant populations is influenced by range size, habitat fragmentation, and population isolation, reflecting the cumulative effects of genetic mutation, genetic drift, gene flow, and natural selection [53]. Previously, Wright et al. [54] established that genetic differentiation among populations can be considered high when Fst values are greater than 0.25, and thus the Fst value of 0.497 we obtained for M. savatieri would tend to indicate a significant genetic differentiation among populations. This assumption was supported by our AMOVA results (P < 0.001), indicating a higher molecular variation among populations (49.74%) than within populations (48.36%). Gene flow is one of the main factors contributing to the maintenance of a homogeneous population genetic structure. Theoretically, at Nm values > 1, there is a lower likelihood of genetic drift, thereby limiting the genetic differentiation among populations, whereas conversely, when Nm < 1, gene flow is insufficient to counteract the effects of genetic drift, thereby favoring an increase in the genetic differentiation among populations [55]. In the present study, we obtained a value of only 0.253 for the gene flow between M. savatieri populations, indicating that low levels of gene exchange would not weaken the differentiation among populations caused by genetic drift, which we suspect could be the primary factor influencing the genetic structure of M. savatieri. It can be reasoned that these findings reflect the characteristics of M. savatieri, such as its semi-parasitism, low reproductive capacity, and low seed viability. Moreover, the viability of M. savatieri populations is increasingly being threatened by habitat fragmentation and destruction, and consequently, declining numbers and hindered gene exchange may lead to a gradual increase in genetic differentiation.
The genetic structure of species reflects the mutation, recombination, genetic drift, and selection effects experienced by populations during the course of evolution [56], and thus population genetic structure analyses are deemed important for gaining an accurate understanding of the genetic relationships among germplasms. In this study, we used three clustering methods (PCoA, STRUCTURE, and UPGMA) to analyze the population structure of M. savatieri. PCoA and UPGMA clustering analyses were performed to classify the assessed materials based on their genetic similarity coefficients and genetic distance, which can clarify the intuitive relationship between populations [57]. However, the results obtained using these two clustering do not provide an indication of the materials that interpenetrated between populations. Conversely, Structure analysis is based on the Hardy-Weinberg equilibrium and Bayesian model algorithm, which eliminates the effects of deviation attributable human factors on population division and can objectively classify populations [58]. Consequently, in the present study, we used these three methods in combination to analyze germplasm resources, thereby enabling us to further characterize the germplasm population structure.
UPGMA clustering analysis divided the 46 individuals collected from 10 different wild regions into two clusters, thereby indicating the presence of two discrete genetic populations among these regions. Using STRUCTURE analysis, these individuals were clearly divided into three different clusters, with a few examples of admixed individuals. These results indicate the relatively homogeneous genetic structure of most samples, and the similar composition of samples collected from the same regions. The results of PCoA analysis were found to be identical to those obtained using STRUCTURE and were in general consistent with the UPGMA clustering tree. In addition, we found that the genetic relationships among populations reflected the natural geographical locations of these populations, which was supported by the Mantel test results, which revealed a significant positive correlation between the geographical and genetic distances among populations (R2 = 0.3304, P < 0.05). Collectively, these results tend to imply that geographical isolation, which would hinder gene exchange between different populations and isolate different gene pools, could be the primary factor contributing to the apparent genetic differentiation among M. savatieri populations.
Conclusions
In the present study, 46 M. savatieri wild individuals were collected from 10 habitats in major distribution provinces in China. Using full-length transcriptome sequencing data, we identified and characterized EST-SSR loci and developed the first set of highly polymorphic EST-SSR markers for M. savatieri. The results of this study indicated that gene penetration among the different M. savatieri populations was relatively low, and the individuals from Zhejiang Province showed the highest genetic diversity. Geographical isolation plays a vital role in genetic differentiation among M. savatieri populations. Based on current trends, effective protection is absolutely necessary and urgent for wild germplasm resources of M. savatieri to maintain their genetic identity and diversity.
Materials and methods
Plant materials and DNA extraction
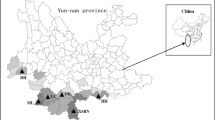
For the purposes of the present study, we collected a total of 46 M. savatieri individuals from different distribution areas in China, among which there were nine accessions from Fujian Province (FJ), 11 from Hunan Province (HN), eight from Jiangxi Province (JX), and 18 from Zhejiang Province (ZJ) (Fig. 7, Supplemental Fig. 2). The formal identification of these plant materials was performed by Prof. Jiankun ** in structured populations. Am J Hum Genet. 2000;7(1):170–81." href="/article/10.1186/s12864-022-08832-x#ref-CR64" id="ref-link-section-d310283e4212">64]. The length of the burn-in period and Markov Chain Monte Carlo (MCMC) run parameters were set to 100,000 iterations. We assessed K values of 1 to 10 based on 10 independent runs for each value. The most favorable K value was determined by estimating the maximum value of the ΔK statistic, using the web-based STRUCTURE Harvester (version 0.9.94) [65], and cluster analysis between populations, based on unweighted pair group with arithmetic average (UPGMA), was performed using NTSYS-pc (version 2.10e) software [66].
Availability of data and materials
All data generated or analysed during this study are included in this article. The sequencing data have been deposited to the CNGB with project accession number CNP0003034 (https://db.cngb.org/search/project/CNP0003034/). The transcript sequences used to detect EST-SSR loci of Monochasma savatieri were deposited and are available at https://github.com/AWan222/LRCyc.
References
Yamazaki T. Scorphulariaceae. In: Iwatsuki K, Yamazaki T, Boufford DE, Ohba H, editors. Flora of Japan, vol. IIIa. Kodansha LTD: Tokyo; 1993. p. 336–41.
Hong D, Yang H, ** C, Holmgren NH. Scrophulariaceae. In: Wu ZY, Raven PH, editors. Flora of China, vol. 18. Bei**g: Science Press and Missouri Botanical Garden Press; 1998. p. 53.
Kohda H, Tanaka S, Yamaoka Y, Yahara S, Nohara T, Tanimoto T, et al. Studies on lens-aldose-reductase inhibitor in medicinal plants. II. Active constituents of Monochasma savatieri Franch. et Maxim. Chen Pharm Bull. 1989;37(11):3153–4.
Fu JX, Wang LX, Wu J. Inflammation Yinning particle treatment of acute upper respiratory tract infection clinical observation. Strait Pharm. 2010;22(10):171–3.
Shi M, He W, Liu Y, Li X, Yang S, Xu Q. Protective effect of total phenylethanoid glycosides from Monochasma savatieri Franch on myocardial ischemia injury. Phytomedicine. 2013;20(14):1251–5.
Zheng W, Tan XQ, Guo LJ, Kong FF, Lu P, Ni DJ, et al. Chemical constituents from Monochasma savatieri. Chin J Nat Med. 2012;10(2):102–4.
Li M, Shi MF, Liu YL, Xu QM, Yang SL. Phenylethanoid glycosides from Monochasma savatieri and their anticomplement activity through the classical pathway. Planta Med. 2012;78(12):1381–6.
Ma J, Xu QM, Wu WQ, Liu CY. Chemical constituents of active fractions of Yanning granules. Chin Tradit Pat Med. 2012;34(10):1946–8.
Yang YN, Wang Y, Sun XB, Kuang HX, Jiang H, Cao JW, et al. Study on anti–inflammatory effects and mechanism of Yanning syrup. Chin Pharm Bull. 2018;34(12):1760–6.
Zhang M, Chen Y, Ouyang Y, Huang Z, Jaime A, Ma G. The biology and haustorial anatomy of semi-parasitic Monochasma savatieri Franch. ex Maxim. Plant Growth Regul. 2015;75:473–81.
Chen X, Guo X, Feng H. Analysis of Phenophase and regeneration characteristics of Monochasma sheareri maxim. J Anhui Agric Sci. 2018;46(27):1–2+38.
Chen L, Zhu Z, Guo Q, Tang S, Huang Z, Zhong W, et al. Growth response of Monochasma savatieri to nitrogen, phosphorus, and potassium in the absence of a host plant: essential demand for nutrients of root hemiparasitic plants. J Plant Nutr. 2020;43(3):444–55.
Environment Agency of Japan. Threatened wildlife of Japan, red data book. Vascular plants. In: Environmental Agency of Japan, vol. 8. 2nd ed. Tokyo: Japan Wildlife Research Center; 2000.
Ouborg NJ. Integrating population genetics and conservation biology in the era of genomics. Biol Lett. 2010;6(1):3–6.
Turchetto C, Segatto AL, Mäder G, Rodrigues DM, Bonatto SL, Freitas LB. High levels of genetic diversity and population structure in an endemic and rare species: implications for conservation. AoB Plants. 2016;8:1–7.
Kim C, Na HR, Choi HK. Genetic diversity and population structure of endangered Isoetes coreana in South Korea based on RAPD analysis. Aquat Bot. 2008;89(1):43–9.
Wu FQ, Shen SK, Zhang XJ, Wang YH, Sun WB. Genetic diversity and population structure of an extremely endangered species: the world’s largest Rhododendron. AoB Plants. 2014;7:10696–700.
Huang LK, Zhang XQ, **e WG, Zhang J, Cheng L, Yan HD. Molecular diversity and population structure of the forage grass Hemarthria compressa (Poaceae) in South China based on SRAP markers. Genet Mol Res. 2012;11(3):2441–50.
Lv J, Qi J, Shi Q, Shen D, Zhang S, Shao G, et al. Genetic diversity and population structure of cucumber (Cucumis sativus L.). Plos One. 2012;7(10):e46919.
Zhao Y, Wang H, Chen W, Li Y, Gong H, Sang X, et al. Genetic diversity and population structure of elite cotton (Gossypium hirsutum L.) germplasm revealed by SSR markers. Plant Syst Evol. 2015;301(1):327–36.
**a W, Luo T, Zhang W, Mason AS, Huang D, Huang X, et al. Development of high-density SNP markers and their application in evaluating genetic diversity and population structure in Elaeis guineensis. Front Plant Sci. 2019;10:130.
Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23(1):48–55.
Singh AK, Chaurasia S, Kumar S, Singh R, Kumari J, Yadav MC, et al. Identification, analysis and development of salt responsive candidate gene based SSR markers in wheat. BMC Plant Biol. 2018;18(1):249.
Parthiban S, Govindaraj P, Senthilkumar S. Comparison of relative efficiency of genomic SSR and EST-SSR markers in estimating genetic diversity in sugarcane. Biotech. 2018;8(3):144.
Poncet V, Rondeau M, Tranchant C, Cayrel A, Hamon S, de Kochko A, et al. SSR mining in coffee tree EST databases: potential use of EST-SSRs as markers for the Coffea genus. Mol Gen Genomics. 2006;276(5):436–49.
Han Z, Ma X, Wei M, Zhao T, Zhan R, Chen W. SSR marker development and intraspecific genetic divergence exploration of Chrysanthemum indicum based on transcriptome analysis. BMC Genomics. 2018;19(1):291.
Guo Q, Wang JX, Su LZ, Lv W, Sun YH, Li Y. Development and evaluation of a novel set of EST-SSR markers based on transcriptome sequences of black locust (Robinia pseudoacacia L.). Genes. 2017;8(7):177.
Yao L, Zheng X, Cai D, Gao Y, Wang K, Cao Y, et al. Exploitation of Malus EST-SSRs and the utility in evaluation of genetic diversity in Malus and Pyrus. Genet Resour Crop Evol. 2010;57(6):841–51.
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177(3):309–34.
Bodénès C, Chancerel E, Gailing O, Vendramin GG, Bagnoli F, Durand J, et al. Comparative map** in the Fagaceae and beyond with EST-SSRs. BMC Plant Biol. 2012;12:153.
Yang B, Xu Q, Niu M, Luo X, Huang H, Tong Z, et al. Analysis of SSR information in transcriptome and development of SSR molecular markers in Rhododendron fortunei. J Nucl Agric Sci. 2018;32(12):2335–45.
Preethi P, Rahman S, Naganeeswaran S, Sabana AA, Gangaraj KP, Jerard BA, et al. Development of EST-SSR markers for genetic diversity analysis in coconut (Cocos nucifera L.). Mol Biol Rep. 2020;47(12):9385–97.
Shelke RG, Basak S, Rangan L. Development of EST-SSR markers for Pongamia pinnata by transcriptome database mining: cross-species amplification and genetic diversity. Physiol Mol Biol Plants. 2020;26(11):2225–41.
Zhang C, Wu Z, Jiang X, Li W, Lu Y, Wang K. De novo transcriptomic analysis and identification of EST-SSR markers in Stephanandra incisa. Sci Rep. 2021;11(1):1059.
Wu J, Cai C, Cheng F, Cui H, Zhou H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol Breed. 2014;34(4):1853–66.
Liu H, Zhang Y, Wang Z, Su Y, Wang T. Development and application of EST-SSR markers in Cephalotaxus oliveri from transcriptome sequences. Front Genet. 2021;12:759557.
Zhang L, Li Y, Tao A, Fang P, Qi J. Development and characterization of 1,906 EST-SSR markers from Unigenes in jute (Corchorus spp.). Plos One. 2015;10(10):e0140861.
Chen C, Xu M, Wang C, Qiao G, Wang W, Tan Z, et al. Characterization of the Lycium barbarum fruit transcriptome and development of EST-SSR markers. Plos One. 2017;12(11):e0187738.
Taheri S, Abdullah TL, Rafii MY, Harikrishna JA, Werbrouck SPO, Teo CH, et al. De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci Rep. 2019;9(1):3047.
Tóth G, Gáspári Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10(7):967–81.
Chai M, Ye H, Wang Z, Zhou Y, Wu J, Gao Y, et al. Genetic divergence and relationship among Opisthopappus species identified by development of EST-SSR markers. Front Genet. 2020;11:177.
Li X, Liu X, Wei J, Li Y, Tigabu M, Zhao X. Development and transferability of EST-SSR markers for Pinus koraiensis from cold-stressed transcriptome through illumina sequencing. Genes. 2020;11(5):500.
Zhou Q, Zhou PY, Zou WT, Li YG. EST-SSR marker development based on transcriptome sequencing and genetic analyses of Phoebe bournei (Lauraceae). Mol Biol Rep. 2021;48(3):2201–8.
Booy G, Hendriks R, Smulders M, Groenendael M, Vosman B. Genetic diversity and the survival of populations. Plant Biol. 2000;2(4):379–95.
Haider N. Chloroplast-specific universal primers and their uses in plant studies. Biol Plant. 2011;5(2):225–36.
Zhu Q, Deng X, Zhang S, Mei S, Chen X, Zhang Y, et al. Genetic diversity of 6 species in Polygonatum by SSR marker. Chin J Chin Mater Med. 2018;43(14):2935–43.
Oh JS, Sa KJ, Hyun DY, Cho GT, Lee JK. Assessment of genetic diversity and population structure among a collection of Korean Perilla germplasms based on SSR markers. Genes Genom. 2020;42(12):1419–30.
Wang SQ. Genetic diversity and population structure of the endangered species Paeonia decomposita endemic to China and implications for its conservation. BMC Plant Biol. 2020;20(1):510.
Wang X, Chen W, Luo J, Yao Z, Yu Q, Wang Y, et al. Development of EST-SSR markers and their application in an analysis of the genetic diversity of the endangered species Magnolia sinostellata. Mol Gen Genomics. 2019;294(1):135–47.
Li XL, Wang J, Fan ZQ, Li JY, Yin HF. Genetic diversity in the endangered Camellia nitidissima assessed using transcriptome-based SSR markers. Trees. 2020;34(2):543–52.
Qiu F, Lei H, Chen J, Yang SC, Bai B, Liu C, et al. EST-SSR analysis of genetic diversity of Psammosilene tunicoides in Yunnan-Guizhou provincial region. Chin Tradit Herb Drugs. 2018;49(16):3895–906.
Wang HX, Hu ZA. Plant breeding system, genetic structure and conservation of genetic diversity. Chin Biodiversity. 1996;4(2):92–6.
Schaal BA, Hayworth DA, Olsen KM, Rauscher JT. Phylogeographic studies in plants: problems and prospects. Mol Ecol. 1998;7(4):465–74.
Wright S. Evolution and the genetics of populations. Variability within and among natural populations, vol. 4. Chicago: University of Chicago Press; 1978. p. 1–30.
Hamriek JL, Godt MJW, Sherman-Broyles SL. Factors influencing levels of genetic diversity in woody plant species. New For. 1992;6:95–124.
Slatkin M. Gene flow and the geographic structure of natural populations. Science. 1987;236(4803):787–9.
Zhang C, Yun L, Zhen LI, Wang J, Guo HY, Sheng Y, et al. Genetic diversity and structure analysis in Psathyrostachys Nevski population using SSR markers. J Plant Genet Resour. 2019;20(1):48–59.
Wei L, Han J, Ren G, Wen F, Wang R, Liu W. Genetic diversity analysis of sesame germplasm resources in Shanxi. Acta Agric Nucl Sin. 2021;35(7):1495–506.
Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;3:411–22.
Li Y, **e Y, Yan T, Liu W, Zhang Y. The research on appraising the purity of maize seeds by utilizing seed albumen polypropylene acyl amine gel electrophoresis. **njiang Agric Sci. 2004;2:125–6.
Cook GA, Prakash O, Zhang K, Shank LP, Takeguchi WA, Robbins A, et al. Activity and structural comparisons of solution associating and monomeric channel-forming peptides derived from the glycine receptor m2 segment. Biophys J. 2004;86(3):1424–35.
Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–9.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics. 2012;28:2537–9.
Pritchard J, Stephens M, Rosenberg N, Donnelly P. Association map** in structured populations. Am J Hum Genet. 2000;7(1):170–81.
Earl DA, Holdt BM. Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Zhang L, Ma Y, Zang DK. Genetic diversity of Stemona shandongensis populations. Bot Res. 2013;2(1):1–5.
Acknowledgments
We thank the local residents for their enthusiastic help during the samples collection. We appreciate for editor and reviewer’s warm work and their valuable comments and suggestions.
Funding
This research was supported by the National Natural Science Foundation of China (31960370), and the Natural Science Foundation of Jiangxi Province, China (20202ACB205002).
Author information
Authors and Affiliations
Contributions
W.Y. and M.Z. performed the experiments. Z.B., F.W. and X.W. performed statistical analyses. W.Y. and F.Z. drafted the manuscript. F.Z. and J.X. contributed to the experimental design and edition of the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All research on the plant materials detailed in this manuscript comply with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora. All plant materials collected in this study are conserved in the greenhouse at Jiangxi Normal University, China, and the seeds are freely available for scientific research. This study was supported by National Natural Science Foundation of China and the Natural Science Foundation of Jiangxi Province, China, including handling these plants and collecting samples.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplemental Table 1.
Details and sources of the 46 Monochasma savatieri samples analyzed in this study. Supplemental Fig. 1. The representative gel pictures with scoring by the EST-SSR markers. The patterns of bands were analyzed by a gel imaging analysis system (version Tocan 240). Supplemental Fig. 2. The photos of some representative samples in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, W., Bai, Z., Wang, F. et al. Analysis of the genetic diversity and population structure of Monochasma savatieri Franch. ex Maxim using novel EST-SSR markers. BMC Genomics 23, 597 (2022). https://doi.org/10.1186/s12864-022-08832-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08832-x