
Abstract
Background
Regulatory circuits of infection in the emerging experimental model system, water flea Daphnia and their microparasites, remain largely unknown. Here we provide the first molecular insights into the response of Daphnia galeata to its highly virulent and common parasite Caullerya mesnili, an ichthyosporean that infects the gut epithelium. We generated a transcriptomic dataset using RNAseq from parasite-exposed (vs. control) Daphnia, at two time points (4 and 48 h) after parasite exposure.
Results
We found a down-regulation of metabolism and immunity-related genes, at 48 h (but not 4 h) after parasite exposure. These genes are involved in lipid metabolism and fatty acid biosynthesis, as well as microbe recognition (e.g. c-type lectins) and pathogen attack (e.g. gut chitin).
Conclusions
General metabolic suppression implies host energy shift from reproduction to survival, which is in agreement with the known drastic reduction in Daphnia fecundity after Caullerya infection. The down-regulation of gut chitin indicates a possible interaction between the peritrophic matrix and the evading host immune system. Our study provides the first description of host transcriptional responses in this very promising host-parasite experimental system.
Similar content being viewed by others
Introduction
Ecological and evolutionary outcomes of host-parasite interactions are receiving renewed attention [1, 2]. It remains challenging to identify model host-parasite systems that are abundant in nature and amenable to experimental manipulation in the laboratory. Such systems are necessary to better understand the mechanistic basis determining the outcome of host-parasite encounters, such as immune response or general metabolic responses of the host. Zooplankton water fleas Daphnia (Crustacea: Cladocera) and their microparasites comprise a promising model system that fulfills these requirements. Daphnia are abundant in standing freshwater bodies and a key component of aquatic food webs as grazers of phytoplankton and a major food source for planktivorous fish and some invertebrates [3]. Daphnia are often infected by a variety of microparasites in nature, including bacteria, fungi, microsporidia and protozoans [4]. Some of these parasites are highly virulent, inducing a strong reduction in host fitness [4, 5] and affecting the outcome of competition between host genotypes [6, 7] or species [8, 9]. Daphnia are easy to maintain in laboratory cultures in conditions where females reproduce asexually via parthenogenesis [4, 10]. Their parasites can also be cultured in vivo in the lab [4, 11].
In the last two decades, Daphnia have been used to study host-parasite interactions [4, 11], becoming one of the 13 model organisms for biomedical research (https://grants.nih.gov/grants/policy/model_organism/). Daphnia have been also used for investigating the evolution of immunity [12, 13], facilitated by an increasing availability of genomic information [14,15,16]. The immune system of arthropods is well described, especially for laboratory model species such as fruit flies Drosophila [17, 18] and mealworm beetles Tenebrio [19], or for vectors of human diseases such as mosquitoes [20]. Some immunity pathways of arthropods are highly conserved between insects and Daphnia [12]. In Daphnia pulex, adaptive evolution in immune system genes is stronger than in non-immune genes [13], as it has also been observed across different Drosophila species under a broad gradient of selective pressures [21]. However, the response of Daphnia immune-related genes to individual parasites is not yet well understood.
Only two studies have assessed the transcriptomic response of Daphnia to parasites, and both used D. magna exposed to the bacterium Pasteuria ramosa. The first study found 45 differentially expressed (DE) genes at 96 h (but not at 40 or 144 h) after parasite exposure; these DE genes were not enriched for Gene Ontology (GO) terms [22]. In the second study, a single immune-related gene, inducible nitric oxide synthase (iNOS), was down-regulated at 4 h (but not at 8 or 12 h) after parasite exposure [23]. The host species in these studies, D. magna, inhabits small, fishless water bodies. Permanent lakes and reservoirs, however, are inhabited by smaller Daphnia species that can withstand fish predation, such as D. galeata [24,25,26]. One of the most abundant parasites of Daphnia species inhabiting European lakes and reservoirs is the gut parasite Caullerya mesnili (Protista: Ichthyosporeans) [27]. C. mesnili causes regular epidemics with prevalence of up to 40% of the entire Daphnia population [6, 28, 29]. C. mesnili can be propagated in vivo in laboratory Daphnia cultures. It spreads horizontally from infected to uninfected Daphnia and a new infection is visible from 8 to 12 days after parasite exposure [30]. Infected Daphnia suffer highly reduced fecundity and survival [27, 31]. Despite the crucial role of C. mesnili in the maintenance of host species [8] and genetic [6] diversity, the mechanisms of how C. mesnili induces host’s defense/adaptive responses to the parasite are unknown.
Here we use Illumina MiSeq RNA-sequencing to produce a transcriptome-wide profile of the response of D. galeata following exposure to the gut parasite C. mesnili, at 4 and 48 h after parasite encounter. We aimed to understand how different components of the Daphnia immune system are recruited and how general metabolic pathways are regulated at a transcriptomic level. We also improved annotation of the D. galeata transcriptome by identifying specific orthologs of immune-related genes to facilitate further studies.
Materials and methods
Host-parasite cultures
Two parasite strains were used in the experiment: i) C. mesnili strain isolated from lake Greifensee (Switzerland) and ii) C. mesnili strain isolated from lake Skulska Wies (Poland). Both strains were isolated in September 2016 (7 months before the experiment was conducted) and separately maintained on a single D. galeata host clone (G100; standard laboratory clone, inbred once) by adding newborns from uninfected stock cultures at approximately 2-week intervals [8]. Both uninfected and infected D. galeata cultures were kept in synthetic Daphnia medium [32], at a constant temperature of 20 ± 1 °C, on a light: dark cycle of 12 h:12 h and with an unlimited food supply of the unicellular green algae Scenedesmus obliquus (> 1 mgL− 1 C, added three times per week).
Experimental setup
We aimed to determine the transcriptional changes of the host D. galeata in response to parasite C. mesnili exposure at two harvesting time points post-exposure (4 and 48 h). To minimise maternal effects, the Daphnia (G100 clone) were cultured for two generations under standardized conditions as described above, except that they were fed daily with 1 mgL− 1 C S. obliquus, and their age and densities were controlled; 10 individuals were raised per jar in 200 mL medium. Adult females (between 21 and 30 days old) were transferred from these stock cultures into six experimental jars containing 200 mL medium each. We standardised the reproductive status of experimental populations, by using 26 gravid and 19 non-gravid females individuals (without eggs or developed ovaries, as checked under a microscope) per jar. Daphnia were fed with 0.5 mgL− 1 C S. obliquus. On the next day, neonates were removed and dead adults were replaced, if necessary. Then, parasite-homogenate and control-homogenate were added (each to three jars). Parasite-homogenate was prepared from G100-individuals heavily infected with C. mesnili. The infected individuals were obtained from stock parasite cultures (34 individuals infected with Greifensee strain and 10 individuals infected with Skulska Wies strain, there was a lower availability of the later strain in laboratory cultures). This or lower parasite/host ratio (i.e. ~ 1 donor Daphnia / 3 recipient Daphnia) has been proven to result in successful infections [8, 33, 34]. Infected Daphnia were homogenized in an Eppendorf tube, using a pestle. The homogenate as well as the medium used to maintain infected Daphnia (infected individuals were selected from stock cultures 24 h beforehand) was equally distributed across the three replicates of parasite-exposed treatment. Control-homogenate was similarly prepared using 44 uninfected hosts and distributed across three replicates of control treatment. After 4 h and then after 48 h post exposure, 11 to 20 Daphnia individuals were collected from each experimental unit. Daphnia were homogenized in an Eppendorf tube with a disposable plastic pestle by hand (~ 5 min), in 150 μl TRIzol reagent (Invitrogen, Carlsbad, USA) at room temperature. A further 850 μl TRIzol reagent was added and the sample was shaken vigorously by hand for 15 s, before being quenched in liquid nitrogen and frozen at − 80 °C.
RNA preparation, library construction and sequencing
Total RNA was extracted from parasite-exposed and control treatments (3 replicates each) at each time point (4 and 48 h post-exposure). 200 μl chloroform was added to each homogenate after thawing. Each sample was shaken vigorously by hand, incubated for 2–3 min at room temperature and then centrifuged at 13,000 g for 15 min at 4 °C. The upper aqueous phase (approx. 540 μl) was transferred to a new RNase-free 1.5 ml microcentrifuge tube and 0.5x volume of absolute ethanol was added to precipitate nucleic acids. The tubes were mixed by inverting 10 times. RNA purification, including on-column DNA digestion, was performed using the RNeasy Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. The quality and quantity of the RNA were determined using a NanoDrop ND-1000 spectrometer (PeqLab, Erlangen, Germany) and an RNA Nano Chip assay on a Bioanalyzer 2100 device (Agilent Technologies, Böblingen, Germany).
Libraries were prepared using the TruSeq Stranded mRNA HT Library Preparation Kit (Illumina, San Diego, USA), according to the manufacturer’s protocol. The libraries were quality checked and quantified using a Bioanalyzer 2100 and a DNA Chip assay. Sequencing was performed on a NextSeq 500 instrument (Illumina, San Diego, USA) as 150-bp paired-end Illumina library. Sequence information was extracted using the CASAVA software (Illumina, San Diego, USA) in FASTQ format. The analysis produced 12 data sets for paired-end sequencing.
Reference transcriptomes
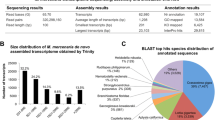
Salmon version 0.8.2 [35] with default parameters was used to pseudo-align raw RNAseq reads to a published D. galeata transcriptome containing 32,903 D. galeata contigs from a mixture of 24 clones isolated from four different lakes [36]. The quality control of this transcriptome was done by Huylmans et al. [36], including the extraction of the longest ORF, the comparison with single copy genes and a MEGAN approach for the presence of non-Daphnia transcripts. To ensure no relevant transcripts were overlooked, a de novo transcriptome was created using RNAseq data produced in this study, resulting in 370,978 contigs (from a single D. galeata clone G100). The de novo transcriptome was assembled using Trinity version 2.4.0 [37] with default parameters. This included quality and adapter trimming using trimmomatic version 0.36 [38] (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:4:5 LEADING:5 TRAILING:5 MINLEN:25) followed by in silico normalization to a maximum coverage of 50. The de novo transcriptome was functionally annotated following the trinotate annotation suite guidelines (https://trinotate.github.io).
Analyses of RNAseq data
After pseudo-alignment of the raw reads with Salmon [35], differentially expressed (DE) transcripts between parasite-exposed and control Daphnia were determined for each time point. This was done for published and de novo reference transcriptome separately, using the R [39] package DESeq2 [40] in conjunction with tximport [41] to aggregate transcript abundances at the gene level. Dispersions were modeled using a local fit with discovery rates at Benjamini-Hochberg-adjusted P-value < 0.05 and absolute value of log2FoldChange > 1. All DE transcripts were translated into amino acid sequences using the Virtual Ribosome package [42] and assigned to eukaryotic orthologous groups (KOGs) with an e-value cut-off of 1e− 5 using the Batch Web CD search tool [43]. Putative orthologs were predicted from reciprocal best BLAST hits. Protein families and signal peptides were identified using Pfam [44] and SignalP [45], respectively. The annotated Pfam domains were transferred to Gene Ontology (GO) terms with Pfam2GO [46].
Only these transcripts that were identified from the published transcriptome were used for GO enrichment analysis. This is because the published transcriptome was quality controlled so redundant contigs were removed [36], whereas the de novo assembly primarily created duplicated contigs. Such redundancies may affect the enrichment analysis as several contigs derived from the same gene would be incorrectly counted. In order to identify over-represented functions (significant at P-value < 0.05) among DE genes, GO enrichment analysis was performed with a hypergeometric distribution using R package GOstats tool [47].
Annotated transcripts (published reference transcriptome) were grouped into manually curated categories based on literature searches highlighting immune functions and based on the information in a Daphnia immunity database [12].
Results
Differentially expressed genes
There were more significantly differentially expressed (DE) genes, as assessed for parasite-exposed vs. control samples, at 48 h than at 4 h post exposure (Fig. 1; the full list of DE genes for each treatment, their accession numbers, P-values and log2FC values are provided in Additional file 1: Table S1). Specifically, after 4 h one gene was down-regulated (detected against the published reference transcriptome), and one up-regulated (detected only against the de novo transcriptome). After 48 h, there were 72 (137) down-regulated and four (31) up-regulated genes, according to the published (Fig. 2) or, in parentheses, de novo transcriptome (Additional file 2: Figure S1). There was no overlap of DE genes between two time points (4 and 48 h).

Volcano plots displaying differential gene expression in Daphnia galeata after exposure to parasite at 4 (left) and 48 (right) hours. Each point represents an individual gene transcript. Red points represent significantly differentially expressed transcripts (Benjamini-Hochberg-adjusted P-value < 0.05)

Venn diagram of the differentially up-regulated (↑) and down-regulated (↓) gene expression in Daphnia galeata at 4 h (solid circle) and 48 h (dashed circle) after parasite exposure. There was no overlap of differentially expressed genes between two time points. Numbers presented here are based on the published reference transcriptome [36], for a similar venn diagram based on the de novo reference transcriptome see Additional file 2: Figure S1
Functional categorization of DE genes
All DE genes were annotated (Additional file 3: Table S2). At the early time point after exposure (4 h), the only up-regulated gene had a putative function of heat shock protein 40 (Hsp40, E-value = 1.45E-15), whereas the only down-regulated gene was not assigned to any putative function (see Additional file 3: Table S2).
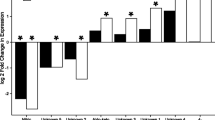
The DE genes identified based on the published transcriptome were used for Gene Ontology (GO) enrichment analysis. The DE genes were enriched for 52 GO terms. These GO terms were classified within three ontologies: cellular component, molecular function, and biological process. After 48 h, eight enriched GO terms were related to lipid metabolism or biosynthesis, and six enriched GO terms were associated with immunity pathways (Fig. 3 and Additional file 4: Table S3).

Gene Ontology (GO) [79] enrichment distributions of Daphnia galeata genes that were down-regulated in parasite-exposed treatment compared to the control treatment at 48 h after parasite exposure. No enriched GO term was identified for the up-regulated genes. Two functional categories are highlighted: metabolism- (green) and immune- (orange) related GO terms
Immune-related genes
Immune-related genes were defined as transcripts encoding putative orthologs or predicted proteins of Daphnia immune system. There were 82 transcripts in the immune protein families that were expressed in the published reference transcriptome. These transcripts were grouped into four main functional classes: pathogen recognition, signal transduction, attack and others (Table 1). A full list of the immune-related genes is provided in Additional file 5: Table S4. Several immune-related genes were down-regulated at 48 h after parasite exposure, such as c-type lectins related to microbe recognition and different chitinases involved in the defense against pathogen attack. Only one immune-related gene, Cytoglobin (Cygb) related to nitric oxide synthase (iNOS) and oxidative stress, was up-regulated at 48 h. At the early stage (4 h), exposure to parasites did not cause a significant change in the expression of immune-related genes (Table 2).
Discussion
The results presented here indicate the existence of coordinated feedbacks occurring between metabolic and immunity pathways in Daphnia galeata in response to Caullerya mesnili infection. This was revealed by changes in gene expression after parasite exposure compared to the uninfected controls. Soon after parasite exposure (4 h), there were very few differentially expressed (DE) genes compared to after 48 h. Only a stress response was triggered in Daphnia as an early action against C. mesnili, whereas the later response resulted in an immune and general metabolic suppression.
Early response
At the early stage (4 h) only a single gene, heat shock protein 40 (Hsp40), was up-regulated. Heat shock proteins are highly conserved proteins and indicators of initial response to different environmental stressors in prokaryotic and eukaryotic organisms [48,49,50]. The changes in the level of heat shock proteins in Daphnia are induced under thermal stress [51], as a reaction to predatory cues [52] or different pollutants [53, 54]. Moreover, an increase of Hsp60 levels was observed in a natural population of Daphnia magna infected by the ectoparasite Amoebidium parasiticum [55]. Apart from the up-regulation of Hsp40 and the down-regulation of one gene not assigned to any putative function, there were no other differentially regulated genes at the early stage after parasite exposure. It might be that the level of threat from the parasite was still too low to trigger the response of the host, or that the host had not yet mounted an immune response specific to C. mesnili attack. A microarray study of D. magna exposed to bacterial endoparasite Pasteuria ramosa showed even more delayed response. In that study, 45 genes were differentially expressed 96 h after exposure, but no differential expression was observed prior (48 h) or after (144 h) [22], which suggests that there is a narrow window in which transcriptomic regulation occurs. It has been proposed that bacterial endoparasites might need some time to penetrate the intestinal lining and colonize the gut epithelium of the Daphnia hosts before the host’s defense mechanisms are activated [4, 56].
Late response
Exposure to parasites resulted in down-regulation of genes involved in chitin metabolism, as assessed 48 h after exposure. Parasites might be causing a weakening of the peritrophic matrix in the Daphnia gut. Chitin metabolism is a fundamental part of arthropod immunity [57], as the peritrophic matrix of arthropod guts forms a protective barrier against ingested pathogens [58, 59]. There are two types of chitin synthases, one responsible for the synthesis of cuticular chitin and the other associated with the gut peritrophic matrix [60, 61]. In our data, we observed a down-regulation of carbohydrate-binding module family 14 (CBM14), also known as peritrophin-A, which is a component of the peritrophic matrix of insect and animal chitinases [62, 63]. In the bumblebee - trypanosome gut parasite system, speritrophin and more genes associated with chitin metabolism were differentially expressed, suggesting an important role for the repair or restructuring of the peritrophic matrix in the host’s response to the parasite [64].
The down-regulation of genes related to c-type lectins at 48 h post exposure implies that C. mesnili could inhibit host cell recognition processes. C-type lectins are involved in the recognition of a variety of pathogens such as fungi, bacteria and viruses [65,66,67]. They bind carbohydrates, and mediate processes of cell adhesion, cell interactions and glycoprotein turnover [66]. In contrast to our finding of down-regulation of c-type lectins, a recent proteomic analysis of D. magna exposed to bacterium P. ramosa revealed their high abundance [68]. However, down-regulation of several c-type lectins was also observed in D. pulex exposed to the predatory phantom midge larvae Chaoborus [69]. Even if the detailed mechanisms of immune responses in Daphnia are still unknown, a threat from either predator or parasite appears to down-regulate c-type lectin expressions.
Two plausible scenarios may explain the observed reduced expression of immune-related genes after parasite exposure (Fig. 4). 1) The parasites may evade host immunity, undermining host defenses for their development in the host’s gut. For example, the gut protozoan parasite Crithidia bombi modifies the immune response of its host bumblebee Bombus terrestris by down-regulating a large number of immune-related genes after infection [64]. Infection of the insect Rhodnius prolixus by the protozoan Trypanosoma cruzi reduces nitric oxide (NO) production which helps the parasite to complete its development in the digestive tract [70]. The principal pathways of innate immunity are conserved between arthropods and vertebrates [71, 72]. A marked down-regulation of immune-related genes was also reported from gilthead sea bream Sparus aurata infected by myxosporean enteric parasite Enteromyxum leei; immune suppressive responses were interpreted as a protection against host tissue damage by degradative enzymes [73, 74]. 2) Down-regulation of a number of metabolism-related genes provides an evidence of general metabolic suppression, including genes involved in lipid metabolism, fatty acid biosynthesis and immune pathways. Under stress conditions, many organisms shift the energy use from reproduction to survival or maintenance of homeostasis according to metabolic cost hypothesis [75,76,77]. Across a diverse array of insects, parasite infections drastically reduced host reproductive output and capacity [78]. This scenario is consistent with the experimental and field observations from D. galeata - C. mesnili system; infected host stops reproducing [28, 31]. Reduced or a completely shut down reproduction seems to be a typical response of Daphnia against various types of microparasites [4].

Schematic cartoon showing changes in immune system and lipid metabolism in Daphnia galeata exposed to gut parasite Caullerya mesnili. Parasite exposure results in down-regulation (red arrows) of immune pathways involved in recognition and attack. At the same time, general metabolic suppression indicates the energy use is shifted from reproduction to survival
Conclusions
We applied an advanced experimental system, the host D. galeata and its gut parasite C. mesnili, to examine the host’s responses on a trascriptomic level. The transcriptome profile of D. galeata after parasite exposure as well as the survey of the immune-related genes of D. galeata have yielded a new example of the parasites’ action to evade the immune defenses of the host, which is critical for disease spread and transmission.
References
Frainer A, McKie BG, Amundsen P-A, Knudsen R, Lafferty KD. Parasitism and the biodiversity-functioning relationship. Trends Ecol Evol. 2018;33(4):260–8.
Penczykowski RM, Laine AL, Koskella B. Understanding the ecology and evolution of host–parasite interactions across scales. Evol Appl. 2016;9(1):37–52.
Lampert W, Sommer U. Limnoecology: the ecology of lakes and streams: Oxford University Press; 2007.
Ebert D. Ecology, epidemiology, and evolution of parasitism in Daphnia. Bethesda: National Library of Medicine (US), National Center for Biotechnology Information; 2005. Available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Books.
Duffy MA. Selective predation, parasitism, and trophic cascades in a bluegill–Daphnia–parasite system. Oecologia. 2007;153(2):453–60.
Turko P, Tellenbach C, Keller E, Tardent N, Keller B, Spaak P, Wolinska J. Parasites driving host diversity: incidence of disease correlated with Daphnia clonal turnover. Evolution. 2018;72(3):619–29.
Capaul M, Ebert D. Parasite–mediated selection in experimental Daphnia magna populations. Evolution. 2003;57(2):249–60.
Wolinska J, Bittner K, Ebert D, Spaak P. The coexistence of hybrid and parental Daphnia: the role of parasites. Proc R Soc B Biol Sci. 2006;273(1596):1977.
Hall SR, Becker CR, Simonis JL, Duffy MA, Tessier AJ, Cáceres CE. Friendly competition: evidence for a dilution effect among competitors in a planktonic host–parasite system. Ecology. 2009;90(3):791–801.
Hebert PDN. The population biology of Daphnia (Crustacea, Daphnidae). Biol Rev. 1978;53(3):387–426.
Ebert D. Host–parasite coevolution: insights from the Daphnia–parasite model system. Curr Opin Microbiol. 2008;11(3):290–301.
McTaggart SJ, Conlon C, Colbourne JK, Blaxter ML, Little TJ. The components of the Daphnia pulex immune system as revealed by complete genome sequencing. BMC Genomics. 2009;10(1):175.
McTaggart SJ, Obbard DJ, Conlon C, Little TJ. Immune genes undergo more adaptive evolution than non-immune system genes in Daphnia pulex. BMC Evol Biol. 2012;12(1):63.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331(6017):555.
Tokishita S-i, Shibuya H, Kobayashi T, Sakamoto M, Ha J-Y, Yokobori S-i, Yamagata H, Hanazato T. Diversification of mitochondrial genome of Daphnia galeata (Cladocera, Crustacea): comparison with phylogenetic consideration of the complete sequences of clones isolated from five lakes in Japan. Gene. 2017;611:38–46.
Bourgeois Y, Roulin AC, Müller K, Ebert D. Parasitism drives host genome evolution: insights from the Pasteuria ramosa–Daphnia magna system. Evolution. 2017;71(4):1106–13.
Leulier F, Parquet C, Pili-Floury S, Ryu J-H, Caroff M, Lee W-J, Mengin-Lecreulx D, Lemaitre B. The Drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat Immunol. 2003;4(5):478.
Kounatidis I, Ligoxygakis P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012;2(5):120075.
Johnston PR, Makarova O, Rolff J. Inducible defenses stay up late: temporal patterns of immune gene expression in Tenebrio molitor. G3: Genes, Genomes, Genetics. 2014;4(6):947–55.
Christophides GK, Vlachou D, Kafatos FC. Comparative and functional genomics of the innate immune system in the malaria vector Anopheles gambiae. Immunol Rev. 2004;198(1):127–48.
Obbard DJ, Welch JJ, Kim K-W, Jiggins FM. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 2009;5(10):e1000698.
Jansen M, Vergauwen L, Vandenbrouck T, Knapen D, Dom N, Spanier KI, Cielen A, De Meester L. Gene expression profiling of three different stressors in the water flea Daphnia magna. Ecotoxicology. 2013;22(5):900–14.
McTaggart SJ, Cézard T, Garbutt JS, Wilson PJ, Little TJ. Transcriptome profiling during a natural host-parasite interaction. BMC Genomics. 2015;16(1):643.
Keller B, Wolinska J, Manca M, Spaak P. Spatial, environmental and anthropogenic effects on the taxon composition of hybridizing Daphnia. Philos Trans R Soc B. 2008;363(1505):2943–52.
Petrusek A, Seda J, Macháček J, Ruthová Š, Šmilauer P. Daphnia hybridization along ecological gradients in pelagic environments: the potential for the presence of hybrid zones in plankton. Philos Trans R Soc B. 2008;363(1505):2931–41.
Yin M, Wolinska J, GießLer S. Clonal diversity, clonal persistence and rapid taxon replacement in natural populations of species and hybrids of the Daphnia longispina complex. Mol Ecol. 2010;19(19):4168–78.
Lohr JN, Laforsch C, Koerner H, Wolinska J. A Daphnia parasite (Caullerya mesnili) constitutes a new member of the Ichthyosporea, a group of protists near the animal–fungi divergence. J Eukaryot Microbiol. 2010;57(4):328–36.
Wolinska J, Keller B, Manca M, Spaak P. Parasite survey of a Daphnia hybrid complex: host-specificity and environment determine infection. J Anim Ecol. 2007;76(1):191–200.
Wolinska J, Seda J, Koerner H, Smilauer P, Petrusek A. Spatial variation of Daphnia parasite load within individual water bodies. J Plankton Res. 2011;33(8):1284–94.
Bittner K, Rothhaupt K-O, Ebert D. Ecological interactions of the microparasite Caullerya mesnili and its host Daphnia galeata. Limnol Oceanogr. 2002;47(1):300–5.
Lohr JN, Yin M, Wolinska J. Prior residency does not always pay off–co-infections in Daphnia. Parasitology. 2010;137(10):1493–500.
Saebelfeld M, Minguez L, Griebel J, Gessner MO, Wolinska J. Humic dissolved organic carbon drives oxidative stress and severe fitness impairments in Daphnia. Aquat Toxicol. 2017;182:31–8.
Tellenbach C, Tardent N, Pomati F, Keller B, Hairston N, Wolinska J, Spaak P. Cyanobacteria facilitate parasite epidemics in Daphnia. Ecology. 2016;97(12):3422–32.
Schoebel CN, Tellenbach C, Spaak P, Wolinska J. Temperature effects on parasite prevalence in a natural hybrid complex. Biol Lett. 2011;7(1):108–11.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417.
Huylmans AK, López Ezquerra A, Parsch J, Cordellier M. De novo transcriptome assembly and sex-biased gene expression in the cyclical parthenogenetic Daphnia galeata. Genome Biol Evol. 2016;8(10):3120–39.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Team RC. R: a language and environment for statistical. Computing. 2013.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Soneson C, Love MI and Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences [version 1; referees: 2 approved]. F1000Research 2015, 4:1521. https://doi.org/10.12688/f1000research.7563.1.
Wernersson R. Virtual ribosome—a comprehensive DNA translation tool with support for integration of sequence feature annotation. Nucleic Acids Res. 2006;34(suppl_2):W385–8.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45(D1):D200–3.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):D222–30.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25.
Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23(2):257–8.
Pauwels K, Stoks R, De Meester L. Co** with predator stress: interclonal differences in induction of heat-shock proteins in the water flea Daphnia magna. J Evol Biol. 2005;18(4):867–72.
Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22(1):631–77.
Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst. 2000;92(19):1564–72.
Mikulski A, Bernatowicz P, Grzesiuk M, Kloc M, Pijanowska J. Differential levels of stress proteins (HSPs) in male and female Daphnia magna in response to thermal stress: a consequence of sex-related behavioral differences? J Chem Ecol. 2011;37(7):670–6.
Pijanowska J, Kloc M. Daphnia response to predation threat involves heat-shock proteins and the actin and tubulin cytoskeleton. Genesis. 2004;38(2):81–6.
Kim H, Kim J-S, Lee Y-M. Changes in activity and transcription of antioxidant enzymes and heat shock protein 90 in the water flea, Daphnia magna - exposed to mercury. Toxicol Environ Heal Sci. 2017;9(5):300–8.
Imhof HK, Rusek J, Thiel M, Wolinska J, Laforsch C. Do microplastic particles affect Daphnia magna at the morphological, life history and molecular level? PLoS One. 2017;12(11):e0187590.
Pauwels K, Stoks R, Decaestecker E, De Meester L. Evolution of heat shock protein expression in a natural population of Daphnia magna. Am Nat. 2007;170(5):800–5.
Duncan AB, Little TJ. Parasite–driven genetic change in a natural population of Daphnia. Evolution. 2007;61(4):796–803.
Beckerman AP, de Roij J, Dennis SR, Little TJ. A shared mechanism of defense against predators and parasites: chitin regulation and its implications for life–history theory. Ecol Evol. 2013;3(15):5119–26.
Kuraishi T, Binggeli O, Opota O, Buchon N, Lemaitre B. Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc Natl Acad Sci. 2011;108(38):15966.
Lehane MJ. Peritrophic matrix structure and function. Annu Rev Entomol. 1997;42(1):525–50.
Arakane Y, Muthukrishnan S, Kramer KJ, Specht CA, Tomoyasu Y, Lorenzen MD, Kanost M, Beeman RW. The Tribolium chitin synthase genes TcCHS1 and TcCHS2 are specialized for synthesis of epidermal cuticle and midgut peritrophic matrix. Insect Mol Biol. 2005;14(5):453–63.
Kumar NS, Tang B, Chen X, Tian H, Zhang W. Molecular cloning, expression pattern and comparative analysis of chitin synthase gene B in Spodoptera exigua. Comp Biochem Physiol B: Biochem Mol Biol. 2008;149(3):447–53.
Shen Z, Jacobs-Lorena M. A type I peritrophic matrix protein from the malaria vector Anopheles gambiae binds to chitin cloning, expression, and characterization. J Biol Chem. 1998;273(28):17665–70.
Elvin CM, Vuocolo T, Pearson RD, East IJ, Riding GA, Eisemann CH, Tellam RL. Characterization of a major peritrophic membrane protein, peritrophin-44, from the larvae of Lucilia cuprina cDNA and deduced amino acid sequences. J Biol Chem. 1996;271(15):8925–35.
Riddell CE, Lobaton Garces JD, Adams S, Barribeau SM, Twell D, Mallon EB. Differential gene expression and alternative splicing in insect immune specificity. BMC Genomics. 2014;15(1):1031.
Weis WI, Taylor ME, Drickamer K. The C–type lectin superfamily in the immune system. Immunol Rev. 1998;163(1):19–34.
Cirimotich CM, Dong Y, Garver LS, Sim S, Dimopoulos G. Mosquito immune defenses against Plasmodium infection. Dev Comp Immunol. 2010;34(4):387–95.
Mayer S, Raulf M-K, Lepenies B. C-type lectins: their network and roles in pathogen recognition and immunity. Histochem Cell Biol. 2017;147(2):223–37.
Otte K: Proteomic analysis of stress responses in Daphnia (Doctoral dissertation). 2016:Retrieved from urn:nbn:de:bvb:19–193402.
Rozenberg A, Parida M, Leese F, Weiss LC, Tollrian R, Manak JR. Transcriptional profiling of predator-induced phenotypic plasticity in Daphnia pulex. Front Zool. 2015;12(1):18.
Castro DP, Moraes CS, Gonzalez MS, Ratcliffe NA, Azambuja P, Garcia ES. Trypanosoma cruzi immune response modulation decreases microbiota in Rhodnius prolixus gut and is crucial for parasite survival and development. PLoS One. 2012;7(5):e36591.
Buchmann K. Evolution of innate immunity: clues from invertebrates via fish to mammals. Front Immunol. 2014;5:459.
Baxter RHG, Contet A, Krueger K. Arthropod innate immune systems and vector-borne diseases. Biochemistry. 2017;56(7):907–18.
Davey GC, Calduch-Giner JA, Houeix B, Talbot A, Sitjà-Bobadilla A, Prunet P, Pérez-Sánchez J, Cairns MT. Molecular profiling of the gilthead sea bream (Sparus aurata L.) response to chronic exposure to the myxosporean parasite Enteromyxum leei. Mol Immunol. 2011;48(15):2102–12.
Pellizzari C, Krasnov A, Afanasyev S, Vitulo N, Franch R, Pegolo S, Patarnello T, Bargelloni L. High mortality of juvenile gilthead sea bream (Sparus aurata) from photobacteriosis is associated with alternative macrophage activation and anti-inflammatory response: results of gene expression profiling of early responses in the head kidney. Fish Shellfish Immunol. 2013;34(5):1269–78.
Rowe CL, Hopkins WA, Zehnder C, Congdon JD. Metabolic costs incurred by crayfish (Procambarus acutus) in a trace element-polluted habitat: further evidence of similar responses among diverse taxonomic groups. Comp Biochem Physiol Part C. 2001;129(3):275–83.
Rokitta SD, Von Dassow P, Rost B, John U. Emiliania huxleyi endures N-limitation with an efficient metabolic budgeting and effective ATP synthesis. BMC Genomics. 2014;15(1):1051.
Lu Y, Wohlrab S, Groth M, Glöckner G, Guillou L, John U. Transcriptomic profiling of Alexandrium fundyense during physical interaction with or exposure to chemical signals from the parasite Amoebophrya. Mol Ecol. 2016;25(6):1294–307.
Schwenke RA, Lazzaro BP, Wolfner MF. Reproduction–immunity trade-offs in insects. Annu Rev Entomol. 2016;61:239–56.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50.
Acknowledgements
We thank Elisabeth Funke for help with the RNA extraction and Nancy Kühne for diligent assistance in the Illumina sequencing at the AWI. Thanks to Mathilde Cordellier and Suda Parimala Ravindran for providing a protocol of RNA extraction and additional information on the published Daphnia transcriptome data. We are grateful to Marcin Dziuba for collecting the parasite Caullerya strain in Poland. We also thank Camila Mazzoni, Susan Mbedi, Felix Heeger, Maximilian Driller, Calvinna Caswara and the entire BeGenDiv crew for their technical support and excellent collaboration. We thank two anonymous reviewers and Michail Kotsyfakis for their comments, which greatly helped improve this manuscript. This work was supported by the joint “lead agency” grant from the German Science Foundation (WO 1587/6-1 to JW) and Swiss National Science Foundation (310030 L 166628 to PS). The publication of this article was funded by the Open Access Fund of the Leibniz Association.
Availability of data and materials
RNAseq raw sequences have been submitted to SRA at NCBI under the Accession no. PRJNA492212. The assembled sequences have been submitted to TSA under the Accession no. GGXW00000000.
Author information
Authors and Affiliations
Contributions
All authors conceived the study. YL and JW performed the experiment. YL carried out the RNAseq library preparation and sequencing under guidance of UJ. YL analyzed the data with help from PJ and wrote manuscript with input from JW and SD. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Full list of Daphnia galeata transcripts that were up- or down-regulated at 4 h and 48 h after parasite exposure (based on published and de novo reference transcriptomes). (XLSX 82 kb)
Additional file 2:
Figure S1. Venn diagram of the differentially up-regulated (↑) and down-regulated (↓) gene expression in Daphnia galeata based on the de novo reference transcriptome at 4 h (solid circle) and 48 h (dashed circle) after parasite exposure. (PNG 46 kb)
Additional file 3:
Table S2. Annotation of all differentially expressed transcripts of Daphnia galeata (based on published and de novo reference transcriptomes). (XLSX 308 kb)
Additional file 4:
Table S3. Gene ontology enrichment of down-regulated genes at 48 h after parasite exposure (based on the published reference transcriptome). (XLSX 60 kb)
Additional file 5:
Table S4. A full list of the immune-related genes in the published reference transcriptome. (TXT 120 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Lu, Y., Johnston, P.R., Dennis, S.R. et al. Daphnia galeata responds to the exposure to an ichthyosporean gut parasite by down-regulation of immunity and lipid metabolism. BMC Genomics 19, 932 (2018). https://doi.org/10.1186/s12864-018-5312-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5312-7