
Abstract
Classification of different cancer types is an essential step in designing a decision support model for early cancer predictions. Using various machine learning (ML) techniques with ensemble learning is one such method used for classifications. In the present study, various ML algorithms were explored on twenty exome datasets, belonging to 5 cancer types. Initially, a data clean-up was carried out on 4181 variants of cancer with 88 features, and a derivative dataset was obtained using natural language processing and probabilistic distribution. An exploratory dataset analysis using principal component analysis was then performed in 1 and 2D axes to reduce the high-dimensionality of the data. To significantly reduce the imbalance in the derivative dataset, oversampling was carried out using SMOTE. Further, classification algorithms such as K-nearest neighbour and support vector machine were used initially on the oversampled dataset. A 4-layer artificial neural network model with 1D batch normalization was also designed to improve the model accuracy. Ensemble ML techniques such as bagging along with using KNN, SVM and MLPs as base classifiers to improve the weighted average performance metrics of the model. However, due to small sample size, model improvement was challenging. Therefore, a novel method to augment the sample size using generative adversarial network (GAN) and triplet based variational auto encoder (TVAE) was employed that reconstructed the features and labels generating the data. The results showed that from initial scrutiny, KNN showed a weighted average of 0.74 and SVM 0.76. Oversampling ensured that the accuracy of the derivative dataset improved significantly and the ensemble classifier augmented the accuracy to 82.91%, when the data was divided into 70:15:15 ratio (training, test and holdout datasets). The overall evaluation metric value when GAN and TVAE increased the sample size was found to be 0.92 with an overall comparison model of 0.66. Therefore, the present study designed an effective model for classifying cancers which when implemented to real world samples, will play a major role in early cancer diagnosis.
Similar content being viewed by others


Introduction
Background of study
The coding part of the genome is referred to as an exome. Any genetic abnormalities in the exomes are known to trigger several types of cancers. With the present prevailing cancer scenario in the world on a constant uprise, extensive research is being carried out to arrive at possible solutions for early diagnosis [1,2,3]. With possible early diagnosis of the disease and application of suitable treatment strategies still hazy in research, there is an urgent need for the design and development of alternative ways that provide faster and precise predictions via comprehending the huge amount of existing cancer data. One important approach is to develop a decision support system (DSS), which predicts patient specific cancer probabilities, and overcomes challenges that arise with wrong treatment decisions and prognosis, massive data interpretation and comprehending patient-specific causes [4]. As an emerging and ever-evolving technology, DSS systems are highly adept at improving the decision-making process, thereby providing support to clinicians and diagnosticians [5]. Currently, there are several approaches to classify the cancer types, based on the exome datasets that are essential for designing a decision support system (DSS) for early diagnosis of cancers [6,7,8,9]. With advent of technology, using artificial intelligence and machine learning on high-throughput data to design an improved DSS model is the premise of the present study.
Related works
Classification algorithms such as support vector machines (SVM), K-nearest neighbors (KNN), Naïve Bayes, decision trees and random forest are primarily being used for cancer classification using machine learning [10, 11]. Studies have previously classified cervical cancer datasets [Contribution of present study The major contribution of our study is towards the development of a highly accurate and improved decision support model, which when used in healthcare, will provide immense benefits to the diagnosis and control of cancers. Additionally, our model encompasses classifications and predictions for five cancer types, making it a novel study with huge potential for early diagnosis of five different cancer types. The reduction of dimensions in the datasets were covered in our study to derive an appropriate derivative dataset which is of utmost importance since they directly contribute to providing better and more accurate predictions on the features of importance. The present study also provides massive insights into the workings of our proposed model, which resulted in a much better overall accuracy when compared to similar such previous work, satisfying the rudimentary aim of our research work, to offer support to the management of healthcare.
Materials and methods
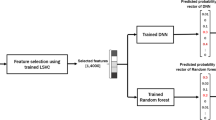
A block diagram summarizing the proposed work, from cleaning and obtaining the derivative exome dataset, using classification analysis by including three classifiers, namely, K-NN, SVM, and a multilayer perceptron network. This was then followed by using majority voting-based ensemble classifier, to finally obtain the proposed results (Fig. 1).

Block diagram summarizing the workflow from cleaning and obtaining the derivative exome datasets, using classification analysis by including three classifiers such as KNN, SVM and a multi-layer perceptron network. This was followed by using majority voting-based ensemble classifiers to obtain the expected results
Dataset analysis
A preliminary analysis of the exome datasets was carried out. These datasets were obtained after a careful analysis of twenty cancer exome datasets, belonging to five cancer types, obtained from our previous work using a standardized workflow (Table 1) [25]. These were human diffuse type gastric cancer, pancreatic adenocarcinoma, high-grade serous ovarian cancer, intrahepatic cholangiocarcinoma, and non-BRCA1/BRCA2 familial breast cancer.
The five cancer types were chosen for our initial analysis in our previous studies because they were the major ones affecting the Indian population, for which we aimed to build a model. Although other cancer types such as hepatocellular carcinoma [29, 30], and bone cancer [31, 32] are also significant, the present study focused on model building for the five types as continuation of our previous work. An extension of this work however, will include more cancer types to stabilise the model further. Additionally, variant identification was also performed in our previous work specifically for these five cancer types which were thought to affect the Indian population more.
Moreover, previous studies have shown that no other similar models were available that were built on these five different cancer types, making our method unique. Please refer Padmavathi et al. [25], for more information on the pipeline used and justifications provided for arriving at different variants. These datasets employed for the study are publicly available and can be downloaded from NCBI SRA (https://www.ncbi.nlm.nih.gov/sra) with their accession numbers.
Hyperlinks for the sample files that were employed in our previous work is provided.
Data clean-up and obtaining a derived dataset
The given exome dataset consisted of 4181 sample variants, with 88 features. On initial analysis, most of the features were filled with NaN (missing value marker). The initial analysis was done using “Pandas” library module available in python modules. These features were dropped, as they couldn’t be used. The features left were 55 in number. These features were still filled with a few of NaN values. Categorical features with NaN values were dropped as well, since these features were not distinct and filling them with the use of Natural Language Processing (NLP) could not have a significant improvement on the precision prediction of the five types of cancer [33]. These included high-grade serous ovarian cancer, pancreatic adenocarcinoma, human diffuse-type gastric cancer, intrahepatic cholangiocarcinoma and non BRCA1/BRCA2 familial breast cancer. Considering only the numerical features for the prediction model, 25 numerical features were obtained. The few NaN values present in the dataset were filled with probabilistic distribution using probabilistic matrix factorization [34]. These 25 features after handling the missing data over the 4181 sample variants constituted the derived dataset.
Exploratory data analysis
Principle Component Analysis (PCA) models were trained over the derived dataset. The number of dimensions in which the dataset was analyzed were one dimension, and two-dimensional axes. The results of the PCA models, reduced the high variance in the dataset due to distributing the weight of the features along two dimensions. Through this distribution the high dimensionality of the dataset was reduced, as the features that would have caused overfitting were removed [35https://colab.research.google.com/drive/1AypJYvigGnpCrhsmLkO6c3b-jSZTqqKN]. The 14 features that had the maximum weight were selected for training the subsequent classification models and were also trained in the ensemble models trained later. The 14 selected features are, ‘shiftscore’ (score for sorting the variants from tolerant to intolerant), ‘TLOD’ (log odds that the variant is present in the tumor sample relative to the expected noise), ‘Sample.AF’ (allelic frequency of the sample), ‘MBQ’ (median base quality of each allele), ‘MFRL’ (median fragment length of each allele), ‘MMQ’ (median map** quality of each allele), ‘Sample.AD’ (allelic depth of the sample), ‘Sample.F1R2’ (forward and reverse read counts for each allele), ‘Sample.F2R1’ (forward and reverse read counts for each allele), ‘DP’ (read depth), ‘GERMQ’ (phred-scaled posterior probability that the alternate alleles are not germline variants), ‘MPOS’ (median distance from the end of the read for each alternate allele), ‘POPAF’ (population allele frequency of the alternate alleles), and ‘Sample.DP’ (approximate read depth of the sample), (https://support.sentieon.com/appnotes/out_fields/) [36]. These parameters provided information on the variants identified from our previous analysis of cancer exomes, with alleles being the alternative forms of the genes that result from mutations and are present on the chromosomes [37]. Since these parameters were found to be most important that could point towards specific cancer types, these were selected for building our model.
This allowed the authors to reduce the bias-variance trade off that would have been caused due to the use of irrelevant features according to the two-dimensional PCA model [38].
Oversampling using SMOTE
Synthetic Minority Oversampling Technique, also referred to as SMOTE, is an oversampling technique to reduce imbalanced datasets. In the exome dataset, it was found that the dataset was heavily imbalanced with the majority class of cancer being Human diffuse-type cancer having the highest number of sample variant (Fig. 2). This would cause the classifiers to not be sensitive to the change in the features of the dataset [39]. In this technique the minority class types to match the number of sample variants in the majority class type were increased using the SMOTE algorithm. This ensured that the imbalance in the dataset was significantly reduced.

Bar graph representing distribution of exome dataset for types of cancer. From the plot, the cancer class for human diffuse type gastric cancer is in majority by a huge margin compared to other classes. This caused the dataset to be imbalanced
Cross validation
Cross validation is a technique used to assess the variance-bias trade-off, of a machine learning model, to understand if the model is overfitting or underfitting, on completely unseen data [40].
The approach followed for cross-validation in our proposed study was hold out cross validation technique. This technique follows by dividing the dataset into a training set and a test set (the test set can be further divided into test and validation set). The model is then trained on the training set, where adjustments are made to its hyper-parameters to balance the variance-bias trade-off. After training the model, the model is subjected to the test set, where all the results produced by the model are considered as a final statement to the performance metrics [40]. This approach was implemented in the present study to cross-validate and confirm the relevance of our model in real-world test scenarios.
K-nearest neighbors classification model analysis
The K-Nearest Neighbors (KNN) machine learning algorithm is an important pattern recognition-based classifier that has great importance in analyzing and predicting cancer types in exome datasets [41, 42]. The primary step in implementing the KNN classifier is to identify the correct number of clusters that the dataset can be divided into. To identify the correct number of clusters, the elbow-curve method was employed. In this method the KNN classifier using the default hyperparameters, for various values of K, i.e., the number of clusters was applied. The order in which the value of K increases is sequential. Then the error rate versus K-graph is plotted. Through this graph the value of K for which the decrement in error rate is the most significant is chosen as the optimal cluster value, K [43]. After obtaining the optimal cluster value, it was used to train the KNN classifier.
The above formula, describes the Euclidean distance method, where N is the dimension of the feature vectors, wk is the dimension of the k-th feature vector, and the pair di and dj, denote the feature vector of a specific text in the training set and the feature vector of another text under consideration in the training set [44].
The default hyperparameters relied on using the Euclidean distance to differentiate the data points into different clusters. This did not result in a better classification. To identify the correct hyperparameters, “Grid Search” module was used [45]. From the grid search module, the best hyperparameters were obtained on training the KNN classifier on different hyperparameters using a verbose of 2. The hyperparameters involved using Manhattan distance, reducing the number of leaf nodes, and using “Ball Tree” algorithm over “Brute Force” algorithm. The classification model was then obtained using these hyperparameters.
For two points (x1,y1), and (x2,y2), the Manhattan distance can be defined as:
where the absolute distance of two points in consideration are calculated. This model is then repeated throughout the different points under consideration for the feature vector present in the dataset, and the classification was carried out [46]. The grid search values are provided in https://colab.research.google.com/drive/1oOBwnfbmy9yLngPSpsJyTCEEOGM_CkmE?usp=sharing#scrollTo=40STvZ9rx8s1 for understanding the range values, which were kept to be a positive integer increment (from 0 to infinity) with verbose of 2.
Support vector machine classification model analysis
Another popular classification model used for data that can be distinguished better with the use hyperplanes and kernel substitution [47]. In this model the Support Vector Machine (SVM) classifier was used with default hyperparameters on the oversampled dataset. The hyperplanes differentiation can be very well implemented for our dataset, due to the high dimensionality [48].
where H represents the hyperplane equation, b is the bias term of the hyperplane equation, and w is the dimension of the feature vector [49].
where the distance function d with reference to a point vector, is given in terms of the symbols defined before [49].
Furthermore, “Grid Search” on SVM classifier using “GridSearchCV” to identify the best hyperparameters on a verbose of 2 was performed, but the results of the “Grid Search” module based on the value ranges as follows,
where ‘C’ is the regularization, which acts as a penalty parameter, ‘gamma’ defines the suitable line of separation, and ‘kernel’(s) are the dimensional modifiers. Within the kernels, ‘rbf’ stands for a Gaussian kernel based on standard normal distribution, and the rest ‘poly’ and ‘sigmoid’ retain their usual meanings.
It was found that the default hyperparameters were best suited for the classification of dataset used in the present study.
Implementing neural networks
Artificial Neural Networks is a complex system that is designed to function and learn like the human brain [50, Full size image

Confusion matrix heatmap of neural network with SMOTE oversampling. The primary diagonal elements from this graph shows the true correct positives and the rest are the false classification. Higher number of primary diagonal from the matrix shows that the classifier has achieved a good accuracy. 0–4 represents the five cancer classes. 0: High-grade serous ovarian cancer, 1: Human diffuse-type gastric cancer, 2: Intrahepatic cholangiocarcinoma, 3: Non BRCA1/BRCA2 familial breast cancer, 4: Pancreatic adenocarcinoma. The light to dark color coding indicates the probabilities of true and false positives

Train-validation accuracy versus epochs and train-validation loss versus epochs for neural network with under sampling. From the graph, it can be seen that the validation accuracy stalls around 40 epochs and has only slight variation after that hence training for 40 epochs should be sufficient to provide same performance as training for 100 epochs. From the validation loss graph, it is noted that after around 40 epochs the model starts to overfit for the training data and hence stop** it after that should prevent it from overfitting

Confusion matrix heatmap of neural network with undersampling. The primary diagonal elements from this graph shows the true correct positives and the rest are the false classification. Higher number of primary diagonals from the matrix shows that the classifier has achieved a good accuracy but performance was worse compared to SMOTE oversampling. 0–4 represents the five cancer classes. 0: High-grade serous ovarian cancer; 1: Human diffuse-type gastric cancer; 2: Intrahepatic cholangiocarcinoma; 3: Non BRCA1/BRCA2 familial breast cancer; 4: Pancreatic adenocarcinoma. The light to dark color coding indicates the probabilities of true and false positives
On the SMOTE oversampled dataset, the individual precision of the above 3 cancers increased significantly; High-grade serous ovarian cancer increased to 0.75, Human Diffuse Type Gastric Cancer increased to 0.83 and Pancreatic adenocarcinoma increased to 0.78. The precision obtained for Intrahepatic cholangiocarcinoma was 0.85 and for Non BRCA1/BRCA2 familial breast cancer was 0.89. This model showed 82.56% validation accuracy after 100 epochs and average accuracy of 82% (Table 2c) on the test set. This model showed to be more stable that the one trained on under sampled dataset and increased the precision and recall for all the types of cancer. Results and codes for the same can be found here https://colab.research.google.com/drive/1lH2tdApkHfqF_6C-d9Pe3o2ZR6oCjp-5, https://colab.research.google.com/drive/1KSDKoxJmbNwW_hBElV2DP-CIlLDA-eP0.
Weighted ensemble learning classifier
As discussed in “Ensemble Machine Learning Approach” section, the base classifiers identified to be ideal had to be weighted according to their performance on the classification of the cancer types. To perform this function, the “tensordot” API available in the “NumPy” module was used (https://numpy.org). The tensordot API helps in calculating the tensor product of the weighted accuracy obtained from the base classifiers. The weighted accuracy of the KNN classifier, SVM classifier, and MLP classifier were 0.754, 0.774, and 0.842 respectively. The ensemble classifier had a weighted accuracy of 82.91% (Table 2d). The dataset was divided into 70:15:15 ratio. The 70:15 was used to split into training and test sets. The remaining 15% was used for the holdout validation set. The performance metric was calculated by fitting the test set to the base classifiers, and then measuring the true positives using majority voting. Using only KNN and SVM classifiers as base classifiers the weighted accuracy of the ensemble estimator still performed better with soft voting, resulting in 78.288%. In this case, the KNN classifier and SVM classifier models had weighted accuracy of 0.736 and 0.701 respectively (https://colab.research.google.com/drive/1mFcOy--VT1hQem8JhClh5TfSK5KnLKJL). The confusion matrix from the resulting ensemble classifier (Table 3), had much better evaluation metrics, with the precision value for high grade serous ovarian cancer, and pancreatic adenocarcinoma reaching 0.76 and 0.83, compared to the results in "Comparison of KNN and SVM classifiers" section. The entire results have been depicted in Table 4, where the performance parameter used for the results is precision. The justification for choosing such a parameter is to allow the weightage of false positives (FP), to have a greater ratio in determining the results as from the statistical relation in "Performance evaluation metrics" section, we observe precision to give us a significant ratio for the same. The weightage of false positives, helps us in the case of prediction of cancer classes based on exome dataset. Precision, has been selected as the required performance metric, as the requirement of having a better ratio in false positives (FP), has a greater significance in cancer prediction for a decision support system. The table therefore, summarizes our proposed models and their respective precision values. The results are presented in SOTA method.
CTGAN and TVAE generated dataset
The proposed model for CTGAN was trained for 300 epochs with a batch size of 10 after which the generator loss was 0.2503 and the Discriminator loss was − 1.4397. The synthetic dataset on evaluation with real dataset with CSTest and KSTest the evaluation metric value was 0.92 and the overall comparison value was 0.66. The proposed TVAE model was also trained for 300 epochs with a batch size of 10 (https://colab.research.google.com/drive/1mFcOy--VT1hQem8JhClh5TfSK5KnLKJL). The synthetic dataset on evaluation with real dataset with CSTest and KSTest, the evaluation metric was 0.93 and the overall comparison value was 0.63.
