
Abstract
Background
Alkaline earth metal ions are important protein binding ligands in human body, and it is of great significance to predict their binding residues.
Results
In this paper, Mg2+ and Ca2+ ligands are taken as the research objects. Based on the characteristic parameters of protein sequences, amino acids, physicochemical characteristics of amino acids and predicted structural information, deep neural network algorithm is used to predict the binding sites of proteins. By optimizing the hyper-parameters of the deep learning algorithm, the prediction results by the fivefold cross-validation are better than those of the Ionseq method. In addition, to further verify the performance of the proposed model, the undersampling data processing method is adopted, and the prediction results on independent test are better than those obtained by the support vector machine algorithm.
Conclusions
An efficient method for predicting Mg2+ and Ca2+ ligand binding sites was presented.
Similar content being viewed by others

Background
The combination of protein and alkaline earth metal ion ligands affects many physiological processes in the human body. For example, vascular smooth muscle must combine with Mg2+ to play the role of dilating blood vessels and regulating blood pressure [1], and thrombin in blood must combine with Ca2+ to perform in coagulation and hemostasis [2]. A large number of studies predicted the binding residues of protein-alkaline earth metal ion ligands. But metal ion ligands are small, active and hard to be predicted, which leads to a generally large false positive in the research results. Therefore, the study of protein-alkaline earth metal ion ligand binding residues is challenging.
Uneven positive and negative data size limits the improvement of prediction accuracy. Generally, there are two kinds of data processing methods: one is to eliminate data imbalance between positive and negative by putting different weight on them. For example, in 2005, Lin et al. [3] used artificial neural network (ANN) method to predict Ca2+ ligand binding residues. In 2016, Hu et al. [14,15,16] of big data. There is a certain similarity between the processing of the natural language problem and the prediction of protein binding residue. Studies showed [17] that performing the frequency analysis on amino acids, their distribution obey Zipf's law, which was considered to be one of the fundamental features of language. This meant that biological sequences can be considered as "natural language" existing in nature and suitable for deep learning algorithms. For example, in 2019, Cui et al. [18] used the Deep convolutional networks algorithm, based on entire amino acid sequences, controlled the size of the effective context scope by the number of convolution layers, and captured the local information of binding residues and the long-distance dependence between amino acids layer by layer to predict the binding residues of six metal ion ligands. While we have processed the protein sequence into shorter amino acid fragments, and controlled the size of the effective context scope. Therefore, we adopted a more concise deep learning algorithm, i.e., deep neural networks (DNN) algorithm. It is built through the fully-connected layers, expresses the essential information contained in the data through multi-layer nonlinear variation, and reduces the dimension of high-dimensional data, so that it can learn more effective features.
Therefore, in this paper, DNN algorithm was used to predict Ca2+ and Mg2+ ligand binding residues, and the results of fivefold cross-validation were better than those of Ionseq method [4] after optimization of hyper-parameters. To further verify the performance of the proposed model, we used the method of undersampling to deal with the data set. By optimizing parameters, we adopted fivefold cross-validation and independent tests. The independent test results were better than those of SVM algorithm. The research showed that: DNN algorithm has certain advantages in predicting Ca2+ and Mg2+ ligand binding residues.
Methods
Establishment of dataset
To ensure the authenticity of the data and the accuracy of the experiment, we selected the data from BioLip database [19] and downloaded protein chain that interacts with Ca2+ and Mg2+ ligands. BioLip database is a semi-manual database, and the data are measured accurately by experiments. To build a non-redundant dataset, we filtered the data and eliminated protein chains with the sequence length of less than 50 amino acids, the resolution of more than 3 Å, and the sequence identity greater than 30%. Compared with Hu et al. [4], the amount of non-redundant data set obtained is obviously increased. The number of protein chains interacting with Ca2+ ligand increases from 179 to 1237, and Mg2+ ligand increases from 103 to 1461.
When a protein combines with a metal ion ligand, both the binding residues and the surrounding residues will be affected. In order to extract more comprehensive information, we used the sliding window method to intercept fragments on protein sequences, and the length L of the intercepted fragments was taken as 9 according to references [6, 7] for the ligands of Ca2+ and Mg2+. To ensure that every amino acid appears in the center of the fragment, we added (L − 1)/2 pseudo amino acids at both ends of the protein chain. If the fragment center was a binding residue, it would be defined as a positive set fragment, otherwise it would be a negative set fragment. The data set of alkaline earth metal ion ligands obtained is shown in Table 1. It can be seen from the data in Table 1 that the fragments of negative set are much larger than those of positive set, the number of fragments of Ca2+ ligand negative set is more than 58 times that of positive set, and that of Mg2+ ligand negative set is more than 92 times that of positive set.
Selection of characteristic parameters
Based on the sequence of amino acids, this paper selected amino acids, physicochemical characteristics of amino acids and predicted structural information as characteristic parameters. Among them, the physicochemical characteristics of amino acids included the charge and hydrophobicity of amino acids. According to the charge properties of amino acids, 20 kinds of amino acids can be divided into 3 categories [20]. Amino acids K, R and H were positively charged, D and E were negatively charged, and other amino acids were not charged. According to the hydrophilic and hydrophobic properties of amino acids, 20 kinds of amino acids were divided into 6 categories [21]. The amino acids R, D, E, N, Q, K and H were strongly hydrophilic, L, I, V, A, M and F are strongly hydrophobic, S, T, Y and W were weakly hydrophilic, and P G and C each belongs to one category. The predicted structural information included secondary structural information, relative solvent accessibility area and dihedral angle (phi angle and psi angle), all of which were obtained from the prediction of protein sequences by the ANGLOR [22] software. The secondary structure information included three types: α-helix, β-fold and random curl. Based on statistical analysis, the area information of solvent accessibility was divided into four intervals [6], x represented the value of relative solvent accessibility area and its threshold was expressed by r(x):
The dihedral angle information was reclassified in line with statistics [13], x represented the angle of the dihedral angle, the threshold value of phi angle was expressed by function g(x), and the threshold value of psi angle was expressed by function h(x):
Extraction of feature parameters
Extraction of component information
We extracted from each fragment for the following component information (37 dimensions):
-
(1)
The frequency of occurrence of amino acids to obtain 21-dimensional amino acid composition information.
-
(2)
The frequency of occurrence of three secondary structures corresponding to amino acids to obtain 4-dimensional secondary structure composition information.
-
(3)
The frequency of 4 relative solvent accessibility area classifications corresponding to amino acids to obtain 5-dimensional relative solvent accessibility information.
-
(4)
The frequency of occurrence of 2 phi angles classifications corresponding to kinds of amino acids to obtain 3-dimensional phi angle component information. Similarly, the psi angle is counted to obtain 4-dimensional psi angle component information.
Conservative characteristics of loci
We used the position weight matrix [23, 24] to extract the conservative features of sites, and the matrix element of the position weight matrix were expressed as follows:
The pseudo-counting probability Pi,j is expressed as:
In the formula, P0,j represents the background probability, and Pi,j represents the occurrence probability of the jth amino acid at the ith site. ni,j represents the frequency of the j amino acid at the i site, Ni represents the total number of amino acids at the i site, and j represents 20 kinds of amino acids and vacancies. q represents the classification number, here 21. Two standard scoring matrices can be constructed from the positive and negative training sets, and each segment can obtain 2L-dimensional feature vectors. Similarly, the predicted secondary structure, relative solvent accessibility area and dihedral angle (phi angle and psi angle) can also be extracted by this method, where q is 4, 5, 3 and 4 respectively.
Finally, we got the information of site conservation in each fragment (2L*5 dimensions):
-
(1)
2L-dimensional position conservation information of 20 amino acids.
-
(2)
2L-dimensional position conservation information of 3 secondary structures.
-
(3)
2L-dimensional position conservation information of 4 relative solvent accessibility.
-
(4)
2*2L-dimensional position conservation information of phi and psi angle.
Information entropy
For the physicochemical characteristics of amino acids, we used information entropy [13, 25] to extract them in order to avoid the information "overwhelming" caused by imbalanced classification.
The information entropy formula is expressed as:
In which pj represents the probability of occurrence of the jth classification in a segment, nj represents the frequency of occurrence of the jth classification in a segment, and N is the segment length. For the value of q, if it represents charge classification, q = 3; if it represents the classification of hydrophilic and hydrophobic, q = 6. Finally, we got the one-dimensional information entropy of hydrophilic and hydrophobic water and the one-dimensional information entropy of charge information.
Deep learning algorithm
Inspired by biological neural network, deep learning algorithm combines low-level features to form a deep neural network with abstract representation, and then simulates the thinking of human brain for perception, recognition and memory, so as to realize high-level feature extraction and expression of complex structural data containing complex information [26]. DNN is one of the common deep learning methods, and its multi-layer network structure expands the neural network's ability to process complex data, processing big data effectively. The protein chain with Ca2+ and Mg2+ ligands contains hundreds of thousands of fragments of positive set and negative set, and its data amount is suitable for deep learning algorithm. Therefore, this paper choosed DNN algorithm as the prediction tool.
The deep learning algorithm modules used in this paper are all implemented in the keras framework:
-
(1)
The normalization module was used to standardize the data to improve the convergence speed and robustness of the training process.
-
(2)
The Earlystop module was used to reduce invalid time cost. If Epoch precision did not rise for 10 consecutive times, it was considered that the best precision has been achieved, and training was stopped to prevent over-fitting.
-
(3)
The Relu function is used as the hidden layer activation function, and the Sigmoid function as the output layer activation function.
Results
Evaluation index
For the evaluation of prediction results, we used the methods commonly used in prediction research of protein-metal ion ligand binding residue [5, 7, 8]: sensitivity (Sn), specificity (Sp), accuracy (Acc), Matthew’s correlation coefficient (MCC). The expressions are:
In which TP represents the number of metal ion ligand binding residues correctly identified; FN represents the number of metal ion ligand binding residues identified as metal ion ligand non-binding residues; TN represents the number of non-binding residues of metal ion ligands correctly identified; FP is the number of metal ion ligand non-binding residues identified as metal ion ligand binding residues.
The prediction results of fivefold cross-validation
Based on the characteristics parameters of secondary structure, relative solvent accessibility area, dihedral angle, charge and hydrophilicity as characteristic parameters, DNN algorithm was used to predict the binding sites. In the results of fivefold cross-validation, the Sn value of Ca2+ and Mg2+ ligands reached 13.1%, Sp and Acc value reached 97.1%, MCC value reached 0.115, and the predicted results were not ideal. Therefore, in order to further improve the prediction accuracy, we optimized the DNN algorithm with hyper-parameters.
Optimization of hyper-parameters
The hyper-parameters of deep learning algorithm include: the number of hidden layers, learning rate, the number of hidden layer nodes and batch sizes, etc. The hyper-parameters has great influence on the training and performance of the model. Therefore, we can optimize the hyper-parameters and select a group of hyper-parameters with the best prediction results, so as to improve the performance of the algorithm. When optimizing a certain kind of hyper-parameters, other hyper-parameters remained unchanged, then the exhaustive method was used in the range of hyper-parameters, and finally a group of parameters with the best prediction performance in the test set was selected. Considering the influence on model accuracy, computing resources and computing time, referring to previous studies [27, 28], we selected three hyper-parameters, namely, the number of hidden layers, the number of hidden layer nodes and the batch size, to optimize, and gave the value range of optimized hyper-parameters, as shown in Table 2.
The impact of changes in the number of hidden layers on the prediction accuracy
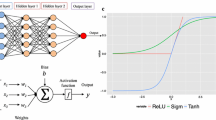
Hidden layer is the network layer between the input layer and the output layer, which has the greatest and most intuitive influence on the network structure, and its number of layers can be adjusted by the feedback of prediction results. Setting the number of hidden layer nodes and batch size as fixed values, the number of hidden layers is increased from 1. The results are shown in Fig. 1.

Curve of MCC value of Ca2+ and Mg2+ ligands with hyper-parameters
Figure 1a is a line chart showing the MCC value of Ca2+ ligand changing with the number of hidden layers. With the increase of the number of layers, the MCC value of Ca2+ ligand gradually increased, and reached the highest point of 0.128 when the number of layers was 3, while the MCC value continued to decrease when the number of layers continued to increase. At the same time, referring to the other three evaluation indexes, it can be determined that the optimal layer value of Ca2+ ligand is 3. Similarly, Fig. 1d is a line chart showing the MCC value of Mg2+ ligand changing with the number of hidden layers. It can be seen from the figure that the optimal layer value of Mg2+ ligand is 5.
The influence of the change of the number of hidden layer nodes on the prediction accuracy
The number of hidden layer nodes need to be adjusted according to the actual situation of the data set. When the number of hidden layer nodes was small, it will be difficult for the network to learn features effectively. Too much hidden layer nodes will increase the complexity of the network structure and reduce the learning speed of the network, which will also lead to over-fitting. Like the process of optimizing the number of hidden layers, we fixed the number of hidden layers and batch size, and then changed the number of hidden layer nodes to find the optimal value. See Fig. 1b, e for the line chart of MCC value changing with the number of nodes. It can be seen that the optimal hidden layer node value of Ca2+ ligand is 32, and Mg2+ ligand is 64.
The impact of batch size changes on prediction accuracy
The value of batch is the number of samples input for training once. Batch size has obvious influence on the data processing and convergence speed of the algorithm. In the previous article, the optimal number of hidden layers and hidden layer nodes had been determined, so we directly optimized the batch size under these two optimal parameters. See Fig. 1c, f for the line chart of MCC value changing with batch size. It can be seen that the optimal batch size of Ca2+ ligand was 32 and Mg2+ ligand was 16.
Finally, we got the optimized hyper-parameters and the optimized prediction results, as shown in Table 3.
In order to verify the reliability and practicability of DNN algorithm, we also compared it with the results of Ionseq method [4], and the results of Ionseq method were also listed in Table 3.
Prediction results based on undersampling method
In order to reduce the influence of data imbalance, we also adopted the method of undersampling [22] to process the data set, and randomly selected the negative sequence fragments equal to the positive set; In order to ensure the stability of the prediction results, the negative set samples were randomly selected 10 times, and the average of the 10 results was taken as the final prediction result. Because the data set constructed by the undersampling method can not accurately simulate the actual forecast situation, we also constructed an independent test data set. The metal ion ligand binding protein chain was divided into two parts: one part accounted for 80% of the total protein chain number, which was used as the training set for the network model, and the other part accounted for 20%, which was used as the independent test set. See Table 4 for the independent test data set of alkaline earth metal ion ligands.
Based on the characteristics parameters of secondary structures, relative solvent accessibility area, dihedral angle, charge and hydrophilic-hydrophobic as characteristic parameters, DNN algorithm was used to predict the binding sites. The results of fivefold cross-validation using training dataset are shown in Table 5. The independent testing dataset was input into the prediction model after the optimization of the hyper-parameter, and the prediction results of independent testing were shown in Table 5.
It can be seen from the results of the fivefold cross-validation in Table 5 that the undersampling method effectively reduces false positives brought by imbalance between positive and negative sets. The Sn values of two ion ligands reach more than 80.1%, and their prediction performance is more balanced. In the results of the independent testing, Sn value of DNN algorithm reaches 71.7%, Sp and Acc value reached 79.1%, MCC value reached 0.163. In order to compare the prediction performance of DNN algorithm in the undersampling method, we compared the results with the results of SVM algorithm using the undersampling method [13], and the prediction results of independent test of SVM algorithm were also listed in Table 5.
Discussion
Comparison in Table 3 shows that the evaluation index of DNN algorithm and Ionseq method had the same characteristics, that is, the Sn value was smaller and the SP value was larger, which was related to the fact that the number of negative sets in the data set was much larger than the number of positive sets. However, Sn value and MCC value of DNN algorithm were better, and Sn value of Mg2+ was 27.2% higher than Ionseq method. The Sp and Acc values of DNN algorithm were slightly lower than those of Ionseq method.
By comparison, it was found in Table 5 that DNN algorithm was better than SVM algorithm except that the Sn value of Mg2+ ligand was slightly lower, and the Sn value of Ca2+ ligand was 11.6% higher than that of SVM algorithm. This may be due to the fact that the number of positive sets of Ca2+ ligands is more than that of Mg2+ ligands, while the DNN algorithm is suitable for big data learning and the SVM algorithm for small sample learning. Therefore, the DNN algorithm has better performance for Ca2+ ligands, and the Sn value of the prediction for Mg2+ ligand was slightly lower. Therefore, based on undersampling method, we think that the prediction performance of DNN algorithm is better than that of SVM algorithm.
Conclusion
In this paper, based on protein sequence information, six characteristic parameters were selected and DNN algorithm was used to predict Ca2+ and Mg2+ ligand binding residues. In order to improve the prediction performance of DNN algorithm, we optimized the number of hidden layers, the number of hidden layer nodes and the batch size of DNN algorithm. With the optimized parameters, the results of fivefold cross-validation were better than those of Ionseq method. At the same time, we also adopted the method of undersampling the data set, and used fivefold cross-validation and independent tests. With the optimized parameters, the independent test results of DNN algorithm were better than those of SVM algorithm. The good prediction results based on the DNN algorithm for predicting Ca2+ and Mg2+ ligand binding residues are due to the large data set of Ca2+ and Mg2+ ligand binding residues, which is suitable for the prediction by the DNN algorithm, and the optimized hyper-parameters of the model, which improves the performance of the algorithm.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DNN:
-
Deep neural network
- ANN:
-
Artificial neural network
- SVM:
-
Support vector machine
- RF:
-
Random forest
- Sp:
-
Specificity
- Acc:
-
Accuracy of prediction
- MCC:
-
Matthew’s correlation coefficient
- Sn:
-
Sensitivity
References
Brailoiu E, Shipsky MM, Yan G, et al. Mechanisms of modulation of brain microvascular endothelial cells function by thrombin. Brain Res. 2016;1657:167–75.
Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52(4):639–72.
Lin CT, Lin KL, Yang CH, et al. Protein metal binding residue prediction based on neural networks. Int J Neural Syst. 2005;15(1–2):71–84.
**uzhen H, Qiwen D, Jianyi Y, et al. Recognizing metal and acid radical ion-binding sites by integrating, ab initio modeling with template-based transferals. Bioinformatics. 2016;32(23):3694–3694.
Jiang Z, Hu XZ, Geriletu G, et al. Identification of Ca2+-binding residues of a protein from its primary sequence. Genet Mol Res. 2016. https://doi.org/10.4238/gmr.15027618.
Cao X, Hu X, Zhang X, et al. Identification of metal ion binding sites based on amino acid sequences. PLoS ONE. 2017;12(8):13.
Wang S, Hu X, Feng Z, et al. Recognizing ion ligand binding sites by SMO algorithm. BMC Cell Biol. 2019;20(Suppl 3):53.
Hu X, Ge R, Feng Z. Recognizing five molecular ligand-binding sites with similar chemical structure. J Comput Chem. 2020;41(2):110–8.
Sodhi JS, Bryson K, McGuffin LJ, et al. Predicting metal-binding site residues in low-resolution structural models. J Mol Biol. 2004;342(1):307–20.
Lin HH, Han LY, Zhang HL, et al. Prediction of the functional class of metal-binding proteins from sequence derived physicochemical properties by support vector machine approach. BMC Bioinform. 2006;7(5):S13.
Horst JA, Samudrala R. Multiple sequence alignment analytic algorithms A protein sequence meta-functional signature for calcium binding residue prediction. Pattern Recogn Lett. 2010;31(14):2103–12.
Lu CH, Lin YF, Lin JJ, et al. Prediction of metal ion-binding sites in proteins using the fragment transformation method. PLoS ONE. 2012;7(6):e39252.
Liu L, Hu X, Feng Z, et al. Recognizing ion ligand-binding residues by random forest algorithm based on optimized dihedral angle. Front Bioeng Biotechnol. 2020;8:493.
Gehrmann S, Dernoncourt F, Li Y, et al. Comparing deep learning and concept extraction based methods for patient phenoty** from clinical narratives. PLoS ONE. 2018;13(2):e0192360.
Lee JK, Choi K, Kim G. Development of a natural language processing based deep learning model for automated HS code classification of the imported goods. J Digit Contents Soc. 2021;22(3):501–8.
Santana LMQD, Santos RM, Matos LN, et al. Deep neural networks for acoustic modeling in the presence of noise. IEEE Lat Am Trans. 2018;16(3):918–25.
Kim JK, Yang SI, Kwon YH, et al. Codon and amino-acid distribution in DNA. Chaos Solitons Fractals. 2005;23(5):1795–807.
Cui Y, Dong Q, Hong D, et al. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinform. 2019;20(1):5.
Jianyi Y, Ambrish R, Yang Z. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2013;2013:D1096–103.
Taylor WR. The classification of amino acid conservation. J Theor Biol. 1986;119(2):205–18. https://doi.org/10.1109/TLA.2018.8358674.
Pánek J, Eidhammer I, Aasland R. A new method for identification of protein(sub)families in a set of proteins based on hydropathy distribution in proteins. Proteins Struct Funct Bioinform. 2005;58(4):923–34.
Wu S, Zhang Y. ANGLOR: a composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS ONE. 2008;3(10):e3400.
Gao S, Hu X. Prediction of four kinds of super secondary structure in enzymes by using ensemble classifier based on scoring SVM. Hans J Comput Biol. 2014;04(1):1–11.
Kel AE, GoBling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCHTM: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;3(1):3576–9.
Strait BJ, Dewey TG. The Shannon information entropy of protein sequences. Biophys J . 1996;71(1):148–55.
Lecun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–44. https://doi.org/10.1038/nature14539.
Cooney C, Korik A, Folli R, et al. Evaluation of hyperparameter optimization in machine and deep learning methods for decoding imagined speech EEG. Sensors. 2020;20(16):4629.
Koutsoukas A, Monaghan KJ, Li X, et al. Deep-learning: investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J Cheminform. 2017;9(1):42.
Acknowledgements
Not applicable.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 22 Supplement 12 2021: Explainable AI methods in biomedical data science. The full contents of the supplement are available at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-22-supplement-12.
Funding
This work was supported by National Natural Science Foundation of China (61961032), Natural Science Foundation of the Inner Mongolia of China (2019BS03025). The funders did not play any role in the design of the study, the collection, analysis, and interpretation of data, or in writing of the manuscript. Publication costs are funded by National Natural Science Foundation of China (61961032).
Author information
Authors and Affiliations
Contributions
KS performed the experiments and wrote the paper. XH and KS analyzed the results, WH, LH and KS participated in the design of the experiment. GZ and ZF gave guidance on the writing of the paper. WZ, SX and YX help on the analysis of the experiments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publication
Not applicable.
Competing interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, K., Hu, X., Feng, Z. et al. Predicting Ca2+ and Mg2+ ligand binding sites by deep neural network algorithm. BMC Bioinformatics 22 (Suppl 12), 324 (2021). https://doi.org/10.1186/s12859-021-04250-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-021-04250-0