
Abstract
Background
The prognosis of wild-type BRAF cutaneous melanoma (WT Bf-CM) patients remains poor due to the lack of therapeutic options. However, few studies have investigated the factors contributing to the prognosis of WT Bf-CM patients.
Methods
In this paper, we proposed and validated a novel 7-RNA based signature to predict the prognosis of WT Bf-CM by analyzing the information from TCGA database.
Results
Dependence of this signature to other clinical factors were verified and a nomogram was also drawn to promote its application in clinical practice. Functional analysis suggested that the predictive function of this signature might attribute to the prediction of the up-regulation of RNA splicing, transcription, and cellular proliferation in the high-risk group, which have been demonstrated to be linked to malignancy of cancer. Moreover, functional analysis and therapy response analysis supported that the prognosis is highly related to PI3K/Akt/mTOR pathway among WT Bf-CM patients.
Conclusion
Collectively, this study will provide a preliminary bioinformatics evidence for the molecular mechanism and potential drug targets that could improving WT Bf-CM prognosis.
Similar content being viewed by others


Introduction
Characterized by rapid progression and metastasis, cutaneous melanoma (CM) is considered as the most aggressive skin cancer, accounting for about 55,500 death worldwide annually [1]. During the past decades, some breakthroughs have been made and significantly improve the survival of metastatic CM patients. For example, the clinical application of BRAF inhibitors and MEK inhibitors, which can extend the progression-free survival (PFS) of BRAF V600-mutate CM patients to 11–14.9 months [1]. However, CM patients carrying BRAF V600 mutations only makes up 45–50% of total melanoma patients [2]. For wild-type BRAF CM (WT Bf-CM) patients, that accounts for about 30% of total CM patients, the response rate to MEK inhibitor is merely 10% [3,4,5]. Lack of therapeutic options resulted in the poor prognosis of WT Bf-CM. With a chemotherapy of dacarbazine, the median PFS of WT Bf-CM patients can be low as 1.5 months [6].
Recently, with the help of next-generation sequencing (NGS) technology, the high heterogeneity of CM has been demonstrated, and it turns out that some features of this heterogeneity such as tumor mutational burden, are associate with the prognosis of CM [7]. Moreover, the high heterogeneity of CM indicates the existence of various biomarkers for prognosis. Although studies have revealed different biomarkers or signature for survival prediction, few of them have focused on the patients with wild-type BRAF CM [8,9,10]. Little is known about which factors can contribute to the poor prognosis of WT Bf-CM patients, apart from less therapy options, failing the need of precise management of WT Bf-CM patients.
To address this issue, this paper analyzed the whole transcription profiles of tumor tissues from 211 patients in the TCGA database, and proposed and validated a novel 7-RNA based signature to predict the prognosis of with wild-type BRAF CM. Moreover, functional analysis and therapy response analysis were also performed, providing a preliminary bioinformatics evidence for the molecular mechanism and potential drug targets that could improving WT Bf-CM prognosis.
Materials and methods
Data extraction
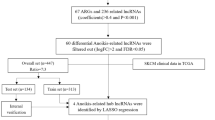
The transcriptional profiles, mutation data and corresponding clinical information of 467 CM patients were downloaded from TCGA-SKCM project with an R package “TCGAbiolinks” [11]. After excluding patients with BRAF somatic mutations and incomplete survival information, 211 WT Bf-CM patients’ tumor samples were equally allocated into training group (n = 106) and testing group (n = 105). NRAS and NF-1 somatic mutational statuses of WT Bf-CM patients were extracted from mutation data. We also downloaded DNA methylation status of specific sites of the WT Bf-CM patients using R package “cgdsr”. RNA-seq data were alignment to the human genome (Gencode.v22 annotation) and the expression of RNAs were measured by Reads Per Kilobase per Million mapped reads (RPKM). RNA expression profiles were identified based on the two following criteria: 1) transcripts were expressed in all WT Bf-CM samples; 2) average of RPKM > 0.1. Eventually, a total of 14,202 RNAs in the profiles were enrolled.
Statistical analysis
In the training set, correlations between the expression of each RNA and overall survival (OS) of patients were calculated by univariate Cox regression and 251 RNAs with their P-values < 0.01 were considered significant to patients’ OS. Random forests were grown based on survival-related RNAs and variable hunting was implemented repeatedly using R package “randomForestSRC”, which output top ranked variables by frequency of occurrence. 7 OS-relevant characteristics were selected as the best and fitted into stepped multivariate Cox regression analysis. Risk scores were weighted by the expression and coefficients of 7 RNAs. The median of risk scores in the training set served as a cut-off, respectively dividing WT Bf-CM patients into high risk groups and low risk groups both in the training set and the testing set. Kaplan–Meier (KM) analysis and visualization were performed to evaluate the survival differences between high risk groups and low risk groups by the “survival” R package. For WT Bf-CM patients with at least 5-year follow ups, receiver operating characteristic (ROC) curves were used to show the sensitivities and specialties of signatures’ abilities to predict the 5-years OS using categorical variables. The area under ROC (AUC) scores and 95% confidence interval (CI) to estimate the reliabilities of signatures were calculated by R package “pROC” [12]. To demonstrate the overview information of risk scores, patients’ survival and expression of signature genes, data were listed in descending order by scaled risk scores and showed through R packages “ggplot2” and “heatmap” as previously described [13]. The results of univariable and multivariable Cox analysis of 7-RNA signature and clinical variables were visualized by the R package “forestplot”.
Nomogram and Decision Curve Analysis (DCA)
Three factors including our signature scores, age and tumor stage which were tightly associated with patients’ survival in multivariable Cox analysis were selected, integrated and used to establish a new model predicting 3-year and 5-year survival of WT Bf-CM patients. Nomogram was drawn with the help of R package “rms”. C-index and calibration curve were used to evaluate the predicting ability of nomogram. DCA is an effective method to estimate molecular signatures and prediction models, especially considering decision preference and clinical demands [14]. Visualization of DCA were achieved in the R statistics environment with the guide of “stdca” (https://github.com/matt-black/dcapy/blob/master/test/resource/stdca.R).
Functional analysis
To explore the function implications of the 7-RNA signature, the differently expression analysis between high risk groups and low risk groups were carried out using R package “edgeR” [15]. Differently expressed genes (DEG) were defined as absolute value of log2FoldChange > 0.8 and adjusted p values < 0.001. The potential interactions of 527 protein-coding DEGs were estimated in String and laid out using Cytoscape [16, 17]. The application in Cytoscape Mcode was used to find the core gene module using criteria of Mcode scores [18]. The top biological process (BP) enrichment results of all DEGs and expression of involved genes were demonstrated and clustered by BP terms using R packages “GOplot” [19]. Gene set enrichment analysis (GSEA) on interested enrichment sets in MSigDB was based on R package “clusterProfiler” [20].
Immune infiltration and drug prediction
To investigate the immune microenvironment of WT Bf-CM tumor samples, 24 sorts of immune cells of each sample were quantified using single sample GSEA (ssGSEA) based on marker genes in previous study and immune infiltration was evaluated by the results of Hierarchical cluster [21, 22]. Combination of immune infiltration results and clinical features was demodnstrated using R package “ComplexHeatmap” [23]. As previously described, we used an R package “pRRophetic” to predict the inhibitory concentration 50 (IC50) of various anti-tumor drugs based on expression profiles [24].
Results
Clinical characteristics of the study populations
A total of 211 WT Bf-CM patients’ samples was downloaded from TCGA. As is shown in Table 1, 64.0% of the samples were collected from male and 36% were from female. The median age of this cohort is 62, and the range of age is 25 ~ 87. 193 samples were matched with pathological stages according to the American Joint Committee on Cancer (AJCC) Cancer staging manual, the percentages of stage 0, I, II, III, IV were 1.9%, 16.6%, 29.9%, 39.3% and 3.8%, respectively. 25.5% of these samples featured with a Breslow thickness < 2 mm, 28.5% within the range of 2 mm to 5 mm, 25.1% > 5 mm. More information of the selected clinical characteristics of are listed in Table 1.
Construction and validation of the expression-based prognostic signature
In order to generate the training set and test set, 211 WT Bf-CM patients were divided equally into two set at random, and their clinical characteristics are shown in Table 1. By analyzing the transcriptome data of training set with univariate Cox regression, 251 RNA were shown significantly to correspond with the 5-year OS of WT Bf-CM patients (p < 0.01). Next, multivariate Cox stepwise regression was performed based on the results of random forests and an expression-based prognostic signature was constructed. This prognostic signature is based on the hazard ratio of 7-RNA and can be formulated as: Risk scores = 0.318*expression of CDC73 + 0.282*expression of RP1-69E11.3 + 0.882*expression of RP11-188D8.1 + 0.968*expression of RP11-116P24.2 + 0.486*expression of TRIB2 + 0.203*expression of VPS13D + 0.199*expression of CELF3. More information about the 7 RNA is listed in Table 2.
The risk scores of each patient from training set were calculated and the median score was used as cut-off for the stratification of high risk group and low risk group. Kaplan–Meier survival analysis and Log-rank test of this two groups showed a significantly poorer prognostics of high risk group compared to low risk group (39.8 months vs. 170.2 months, p < 0.001) (Fig. 1a). For the prognostic value of the proposed signature, ROC curve of training set was drawn and AUC was calculated to be 0.866 (Fig. 1a). To further validate the performance of the proposed signature, patients from test set were stratified similarly based on the median score from training set. Same analysis and tests were also performed with the high- and low-risk groups of test set. Besides the significant correspondence between risk-score and prognosis (59.3 months vs. 90.4 months, p = 0.005), the ROC curve (AUC = 0.699) reveals the considerable potential of this signature for predicting the prognosis of WT Bf-CM patients in the test set (Fig. 1b). Similar results were also observed in the entire WT Bf-CM set that high risk group had shorter OS than low risk group (47.9 months vs. 170.2 months, p < 0.001) and the AUC was 0.780 (Fig. 1c). Risk-related profiles revealed that more deaths occurred and expression of signature genes increased with the raise of risk scores (Fig. 1d).

The 7-RNA signature-related risk score and OS prediction of WT Bf-CM patients. KM analysis reveal the OS differences between high and low risk groups and ROC curves assess the predictive performance of the signature-related score in training set (a); in testing set (b) and in the entire set (c). P values were calculated by two-sided log-rank tests, total AUC values were estimated and 95% CI were computed with 2000 stratified bootstrap replicates. d Risk-related plots illustrate the risk scores, survival status of patients and heatmap of 7 RNAs’ expression
Independence evaluation of the signature in prognostic prediction from clinical and pathological factors
Traditional clinical and pathological factors have been well-clarified to be associated with the prognosis of melanoma patients, including age, gender, pathological stage, Breslow depths and radiation therapy [1, 25]. Using univariable and multivariable analysis, age and stage were shown to be significantly associated with the prognosis of 211 WT Bf-CM patients in addition to the proposed signature (Fig. 2a). Therefore, age and stage were chosen to be compared with the proposed signature for its independence. As is shown in Fig. 2b, when applied in each subgroup of entire set divided according to the risk stratification of each factor, the proposed signature revealed considerable performances in prognosis prediction. These results support the independence of this expression-based prognostic signature. Finally, we also examined the performances of our signature in CM considering less common mutations including NF-1 mutant, NRAS-mutant, and triple negative CM [26]. The results are shown in Fig. S1 and further support that our 7-RNA signature was independent of NRAS and NF-1 mutant status in BRAT-WT CM.

Signature’s independence of clinical factors and stratification analysis. Univariable Cox regression and multivariable Cox regression analysis were performed, P-value (significance), Hazard Ratio and 95% CI were respectively shown in (a) and (b). KM and ROC analysis of regrou** cohorts based on age and stage were correspondingly demonstrated in (c) and (d)
Comparison of the signature with other known prognostic biomarkers
Considering the burden brought by CM, several studies have proposed other biomarkers for CM prognosis prediction [8, 27,28,29,30,31]. Although none of them specifically aimed at WT Bf-CM, we compared our signature with them to examine the predictive performances in WT Bf-CM prognosis. As is shown in Fig. 3a and Table 3, the ROC analysis of time-dependent analysis in WT Bf-CM cohort proved that our signature had significant higher AUC than other biomarkers, and might be superior in WT Bf-CM prognosis prediction exclusively.

a Comparison of predictive performances of our 7-RNA signature and other known biomarkers/signatures. b The signature-based nomogram to predict 3-year and 5-year survival probability. c The calibration plot of nomogram predicted and real surviving proportions. d Net reduction interventions and (e) net benefit of decision curve analysis
Construction and evaluation of a prognostic nomogram for WT Bf-CM
Nomogram has been a welcomed quantitative predictive tool for clinical practice. Based on the results from univariable and multivariable analysis above (Fig. 2a), a nomogram to predict 3 − year and 5-year survival of WT Bf-CM patients was constructed based on the risk scores of the proposed signature, age and pathological stage (Fig. 3b). The C-index of this nomogram was calculated to be 0.7505 and calibration plot was also drawn, exhibiting an acceptable accuracy for prediction (Fig. 3c). Additionally, DCA was performed to assess the efficiency of the proposed nomogram (Fig. 3d, e). When the threshold probability was lower than 25%, our nomogram would be more beneficial and when threshold probability higher than 25%, our signature would be more beneficial. These results supported the promising complementary use of our signature and nomogram.
Functional analysis based on the signature grou**
Cellular and molecular mechanism accounting for the prognosis of melanoma have always been a hot topic of cancer research. Thus, we aimed to find out what biological process might explain the predictive function of this signature. 865 genes were defined as DEGs between high risk and low risk group (Fig. 4a) and interactions of 527 proteins provided a landscape of potential molecular mechanisms (Fig. 4b). Using Mcode, a six-gene module with highest score (score 6) was selected as core module, which was closely associated with mRNA processing, splicing and metabolic process (Fig. 4b). BP enrichment analysis showed various processes were involved, including response to oxygen levels, RNA splicing, regulation of developmental growth, etc. (Fig. 4c). Besides, GSEA analysis indicated that the high-risk group was associated with the up-regulation of GO_NUCLEAR_SPECK (NES = 1.361, p = 0.026), GO_PYRIMIDINE_NUCLEOTIDE_BIOSYNTHETIC_PROCESS (NES = 1.573, p = 0.039), REACTOME_MITOTIC_PROMETAPHASE (NES = 1.413, p = 0.041) and TATA_01 (NES = 1.589, p = 0.005) (Fig. 4d). Taken together, these results supported that the predictive function of this signature might be related to the up-regulation of RNA splicing, transcription, and cellular proliferation in the high-risk group, and upregulation of these processes have been demonstrated to be linked to malignancy of cancer [32, 33].

a Volcano plot of DEGs between high risk and low risk group. b Protein–protein interactions (PPI) of DEGs in String and core module of PPI with the highest Mcode score. c GOplot of BP enrichment results showing term-clustered gene set and fold changes of genes. d GSEA results of specific enrichment set in MSigDB
Evaluation for the therapeutic responses based on signature grou**
The treatment of cutaneous melanoma has made a considerable progress recently, especially for the application of immune-therapy and targeted drugs. Hence, we wondered if the proposed signature could provide clues for the therapeutic responses of WT Bf-CM patients. By estimating the immune infiltration of the WT Bf-CM cohort, we found that little differences in 24 sorts of immune cells infiltration (Fig. 5a). Similarly, the comparison of the expression of 9 immune-markers showed little differences between high- and low- risk groups (Fig. 5b). Of note, although the checkpoint therapies targeting programmed cell death 1 (PD-1), programmed cell death-ligand 1 (PD-L1) and cytotoxic T lymphocyte antigen 4 (CTLA-4) have been recommended for the treatment of WT Bf-CM, their expression didn’t seem to be high both in high- or low-risk groups, suggesting the poor responses of checkpoint therapy in WT Bf-CM patients. Interestingly, the analysis of responses to chemo-therapies showed significant differences of 9 chemotherapeutic drugs in estimated IC50 between high and low-risk groups, along with an increased IC50 to these 9 chemotherapies in high risk group (Fig. 5c). Among these 9 chemotherapeutic drugs, the targets of AKT.inhibitor.VIII, AS601245, GDC0941, MK.2206, PF.02341066 (Crizotinib), Rapamycin and Temsirolimus are involved in the regulation of PI3K/Akt/mTOR pathway [34, Collectively, this study proposed a novel 7-RNA signature for the prognostic prediction of WT Bf-CM. As far as we are concerned, this is the first prognostic signature for WT Bf-CM, which might provide more assistance in the management of WT Bf-CM patients.Conclusion
Availability of data and materials
All the data can be downloaded from https://portal.gdc.cancer.gov/.
Abbreviations
- WT Bf-CM:
-
Wild-type BRAF cutaneous melanoma
- CM:
-
Cutaneous melanoma
- PFS:
-
Progression-free survival
- NGS:
-
Next-generation sequencing
- RPKM:
-
Reads Per Kilobase per Million mapped reads
- OS:
-
Overall survival
- ROC:
-
Receiver operating characteristic
- AUC:
-
The area under ROC
- DCA:
-
Decision curve analysis
- DEG:
-
Differently expressed genes
- GESA:
-
Gene set enrichment analysis
- BP:
-
Biological process
- IC50:
-
Inhibitory concentration 50
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death-ligand 1
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
References
Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S. Melanoma. The Lancet. 2018;392(10151):971–84.
Verduzco D, Kuenzi BM, Kinose F, Sondak VK, Eroglu Z, Rix U, Smalley KSM. Ceritinib Enhances the Efficacy of Trametinib in BRAF/NRAS-Wild-Type Melanoma Cell Lines. Mol Cancer Ther. 2018;17(1):73–83.
Capparelli C, Purwin TJ, Heilman SA, Chervoneva I, McCue PA, Berger AC, Davies MA, Gershenwald JE, Krepler C, Aplin AE. ErbB3 Targeting Enhances the Effects of MEK Inhibitor in Wild-Type BRAF/NRAS Melanoma. Cancer Res. 2018;78(19):5680–93.
Frankel AE, Eskiocak U, Gill JG, Yuan S, Ramesh V, Froehlich TW, Ahn C, Morrison SJ. Digoxin Plus Trametinib Therapy Achieves Disease Control in BRAF Wild-Type Metastatic Melanoma Patients. Neoplasia. 2017;19(4):255–60.
Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, Sun P, Moy C, Szabo SA, Roadcap LT, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9.
Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, Rutkowski P, Del Vecchio M, Gutzmer R, Mandala M, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(4):435–45.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017;16(11):2598–608.
Guo W, Zhu L, Zhu R, Chen Q, Wang Q, Chen JQ. A four-DNA methylation biomarker is a superior predictor of survival of patients with cutaneous melanoma. Elife. 2019;8:e44310.
Jiang Y, Shi X, Zhao Q, Krauthammer M, Rothberg BE, Ma S. Integrated analysis of multidimensional omics data on cutaneous melanoma prognosis. Genomics. 2016;107(6):223–30.
Yang S, Xu J, Zeng X. A six-long non-coding RNA signature predicts prognosis in melanoma patients. Int J Oncol. 2018;52(4):1178–88.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8): e71.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77.
Li X, Shi Y, Yin Z, Xue X, Zhou B. An eight-miRNA signature as a potential biomarker for predicting survival in lung adenocarcinoma. J Transl Med. 2014;12:159.
Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med Inform Decis Mak. 2008;8:53.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2.
Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–4.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95.
Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9.
Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE. 2014;9(9): e107468.
Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Bastholt L, Bataille V, del Marmol V, Dréno B, Fargnoli MC, et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics – Update 2019. Eur J Cancer. 2020;126:141–58.
Eggermont AM, Spatz A, Robert C. Cutaneous melanoma. Lancet. 2014;383(9919):816–27.
Chen X, Guo W, Xu XJ, Su F, Wang Y, Zhang Y, Wang Q, Zhu L. Melanoma long non-coding RNA signature predicts prognostic survival and directs clinical risk-specific treatments. J Dermatol Sci. 2017;85(3):226–34.
Shi G, Li H, Gao F, Tan Q. lncRNA H19 predicts poor prognosis in patients with melanoma and regulates cell growth, invasion, migration and epithelial-mesenchymal transition in melanoma cells. Onco Targets Ther. 2018;11:3583–95.
Timani KA, Gyorffy B, Liu Y, Mohammad KS, He JJ. Tip110/SART3 regulates IL-8 expression and predicts the clinical outcomes in melanoma. Mol Cancer. 2018;17(1):124.
Wang Q, Wang X, Liang Q, Wang S, Liao X, Li D, Pan F. Prognostic Value of Dynactin mRNA Expression in Cutaneous Melanoma. Med Sci Monit. 2018;24:3752–63.
Zhang W, Li X, Song G, Luo D. Prognostic significance of LKB1 promoter methylation in cutaneous malignant melanoma. Oncol Lett. 2017;14(2):2075–80.
Dong X, Chen R. Understanding aberrant RNA splicing to facilitate cancer diagnosis and therapy. Oncogene. 2020;39(11):2231–42.
Loberg RD, Bradley DA, Tomlins SA, Chinnaiyan M, Pieta KJ. The lethal phenotype of cancer: The molecular basis of death due to malignancy. Ca-Cancer J Clin. 2007;57(4):225–41.
Hu N, Zhang Y. TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-kappaB/JNK-dependent activation of autophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2001–11.
Li J, **ao Q, Bao Y, Wang W, Goh J, Wang P, Yu Q. HER2-L755S mutation induces hyperactive MAPK and PI3K-mTOR signaling, leading to resistance to HER2 tyrosine kinase inhibitor treatment. Cell Cycle. 2019;18(13):1513–22.
Ji S, Lin W, Wang L, Ni Z, ** F, Zha X, Fei G. Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. J Cancer. 2017;8(4):555–62.
Davis WJ, Lehmann PZ, Li W. Nuclear PI3K signaling in cell growth and tumorigenesis. Front Cell Dev Biol. 2015;3:24.
White RJ, Sharrocks AD. Coordinated control of the gene expression machinery. Trends Genet. 2010;26(5):214–20.
Ny L, Hernberg M, Nyakas M, Koivunen J, Oddershede L, Yoon M, Wang X, Guyot P, Geisler J. BRAF mutational status as a prognostic marker for survival in malignant melanoma: a systematic review and meta-analysis. Acta Oncol. 2020;59(7):833–44.
Zheng HC, Gong BC, Zhao S. The clinicopathological and prognostic significances of CDC73 expression in cancers: a bioinformatics analysis. Oncotarget. 2017;8(56):95270–9.
Wang J, Zuo J, Wahafu A, Wang MD, Li RC, **e WF. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci Ther. 2020;26(3):297–308.
Hill R, Kalathur RK, Colaco L, Brandao R, Ugurel S, Futschik M, Link W. TRIB2 as a biomarker for diagnosis and progression of melanoma. Carcinogenesis. 2015;36(4):469–77.
Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, Dolinski B, Kraus M, Roberts B, Arthur W, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med. 2010;2(43):43ra55.
Zhou B, Guo R. Integrative analysis of significant RNA-binding proteins in colorectal cancer metastasis. J Cell Biochem. 2018;119(12):9730–41.
Falkenius J, Johansson H, Tuominen R, FrostvikStolt M, Hansson J, EgyhaziBrage S. Presence of immune cells, low tumor proliferation and wild type BRAF mutation status is associated with a favourable clinical outcome in stage III cutaneous melanoma. BMC Cancer. 2017;17(1):584.
Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, Lazar AJ, Faries MB, Kirkwood JM, McArthur GA, et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67(6):472–92.
Kohn CG, Zeichner SB, Chen Q, Montero AJ, Goldstein DA, Flowers CR. Cost-Effectiveness of Immune Checkpoint Inhibition in BRAF Wild-Type Advanced Melanoma. J Clin Oncol. 2017;35(11):1194–202.
Jessurun CAC, Vos JAM, Limpens J, Luiten RM. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front Oncol. 2017;7:233.
Kumpers C, Jokic M, Haase O, Offermann A, Vogel W, Gratz V, Langan EA, Perner S, Terheyden P. Immune Cell Infiltration of the Primary Tumor, Not PD-L1 Status, Is Associated With Improved Response to Checkpoint Inhibition in Metastatic Melanoma. Front Med (Lausanne). 2019;6:27.
Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–76.
Zhang JH, Seigneur EM, Pandey M, Loshakov A, Dagur PK, Connelly PS, Koo L, Panicker LM, Simonds WF. The EIF4EBP3 translational repressor is a marker of CDC73 tumor suppressor haploinsufficiency in a parathyroid cancer syndrome. Cell Death Dis. 2012;3:266.
Hill R, Madureira PA, Ferreira B, Baptista I, Machado S, Colaco L, Dos Santos M, Liu N, Dopazo A, Ugurel S, et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat Commun. 2017;8:14687.
Acknowledgements
We particularly thank the support and care in academic spirits from Di Fu (Shanghai Jiao Tong University School of Medicine) and Qi Qin (Sun Yat-sen University Zhongshan School of Medicine).
Code availability
All the code can be guided by manuscripts of corresponding R packages.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Data collection: R.S.,L.L.; study design: Z. T.,L.L.; writing: Z. T., C.L and R. S; data analysis: R.S., Y. L and Z.T.,L.L.; data interpretation/revision/final approval: all authors. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
No ethics approval should be declared.
Consent for publication
All the involved members agreed for publication.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
KM analysis reveal the OS differences between high and low risk groups in NF-1 mutant (a), NRAS-mutant (b), and triple-wild-type (c) WT Bf-CM patients. ROC curves in corresponding subgroups (d-f) assess the predictive performance of the signature-related score. P values were calculated by two-sided log-rank tests, total AUC values were estimated and 95% CI were computed with 2000 stratified bootstrap replicates.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, R., Liu, Y., Lei, C. et al. A novel 7 RNA-based signature for prediction of prognosis and therapeutic responses of wild-type BRAF cutaneous melanoma. Biol Proced Online 24, 7 (2022). https://doi.org/10.1186/s12575-022-00170-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12575-022-00170-2