
Abstract
Background
The mining of new allelic variation and the induction of new genetic variability are the basis for improving breeding efficiency.
Results
In this study, in total, 3872 heavy ion-irradiated M2 generation rice seeds and individual leaves were collected. The grain length was between 8 and 10.22 mm. The grain width was between 1.54 and 2.87 mm. The results showed that there was extensive variation in granulotype. The allelic variation in GS3 and GW5 was detected in 484 mixed samples (8:1) using targeted sequencing technology, and 12 mixed samples containing potential mutations and 15 SNPs were obtained; combined with Sanger sequencing and phenotype data, 13 key mutants and their corresponding SNPs were obtained; protein structural and functional analysis of key mutants screened out 6 allelic variants leading to altered grain shape, as well as the corresponding mutants, including long-grain mutants GS3-2 and GS3-7, short-grain mutants GS3-3 and GS3-5, wide-grain mutant GW5-1 and narrow-grain mutant GW5-4; whole genome sequencing identified new grain length gene allelic variants GS3-G1, GS3-G2 and GS3-G3.
Conclusion
Based on the above studies, we found 6 granulotype mutants and 9 granulotype-related allelic variants, which provided new functional gene loci and a material basis for molecular breeding and genotype mutation and phenotype analysis. We propose a method for targeted identification of allelic variation in rice grain type genes by combining targeted sequencing of mixed samples and whole genome sequencing. The method has the characteristics of low detection cost, short detection period, and flexible detection of traits and genes.
Similar content being viewed by others

Background
Rice (Oryza sativa L.) is one of the three major food crops and is a staple food for nearly half of the world’s population (Miura et al. 2010). Excellent germplasm resources are the basis for improving the breeding efficiency of new rice varieties (Zhao et al. 2011). Various physical or chemical mutagenic factors can induce changes in biological genetic material, resulting in new allelic variations and species. As a new radiation mutagenesis method, heavy ion mutagenesis has unique advantages, such as a high mutation rate, wide mutation spectrum, fast mutation stability, and stable and reliable mutagenesis, and this method is simple and easy to implement (Qu et al. 2007; Hase et al. 2012). Due to the high linear energy transfer (LET) properties of heavy ion beams, single nucleotide variations (SNVs) and insertions/deletions (InDels) and structural variations (SVs) can be induced at higher frequencies (Zheng et al. 2021). This method can induce heritable variation in plant genomes in contemporary times. The resulting mutants are important materials for functional genomics research (Oono et al. 2020). Heavy ion radiation is one of the effective ways to innovate rice germplasm (**g et al. 2021). In recent years, this technique has played an important role in plant breeding (Yang et al. 2019a; Li et al. 2019; Sjahril et al. 2020; Okasa et al. 2021; Zhang et al. 2022).
Mining of allelic variations is the key to creating new germplasm for plants and animals. The rice Wx gene is the main gene that controls amylose synthesis, and the discovery and utilization of its allelic variation is an important way to analyze rice quality variation and is also an important basis for rice quality improvement. Currently, the Wx gene has been discovered and identified, and multiple important allelic variants, including Wxlv, Wxa, Wxb, Wxin, Wxmp, Wxop, and wx (Zhang et al. 2019), have also been identified. GS3 is a major QTL controlling grain length in rice, and the protein it encodes negatively regulates grain length (Fan et al. 1997; Zhang et al. 2012; Fazio et al. 2003). The targeted induced local lesions in genomes (TILLING) technique is a reverse genetics technique developed in the 1990s. This technique is based on chemical mutagenesis materials and combines chemical mutagenesis technology with PCR screening technology and high-throughput detection methods. Linked together, a high-throughput and rapid detection of point mutations in target gene regions has formed a technical system (Henikoff and Comai 2003) that has been applied in a variety of plants and promoted mutagenesis and breeding development (Boualem et al. 2014; Anai 2012; Ochiai et al. 2011; Chen and Dubcovsky 2012; Minoia et al. 2010). MutMap is a forward genetic gene map** strategy and genetic analysis method developed based on whole genome sequencing (WGS) (Abe et al. 2012). The MutMap method also includes a variety of developments and extensions, such as MutMap+ and MutMap-Gap. These methods do not require the establishment of cumbersome progeny map** groups and do not rely on genetic hybridization and any linkage information. The identification process of variant loci has been successfully applied to the study of multiple gene map**s in different species (Takagi et al. 2013a, b, 2015; Rym et al. 2017). In recent years, targeted sequencing technology has been widely used. Targeted sequencing is GenoPlexs based on multiplex PCR and GenoBaits based on liquid-phase probe capture, which can detect multiple SNPs within a single amplicon, greatly improving intratarget variation and detection efficiency. This technique has the characteristics of high marker flexibility and high detection efficiency and can be widely used in biological evolution, genetic map construction, gene location cloning, marker trait association detection, allelic variation detection, etc. (Shen et al. 2021; Guo et al. 2019; Lu et al. 2019; Li et al. 2020; Du et al. 2019; Yang et al. 2019b).
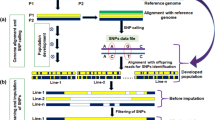
In this study, 3872 12C6+ radiation mutagenized mutant materials for the second generation were identified by mixed sample targeted sequencing technology, and the mutant mixed samples and SNPs related to the granulotype genes GS3 and GW5 were mined and then selected by Sanger sequencing. Mutant individual plants, combined with phenotype and protein function analysis, were utilized to further select key mutant individual plants and conduct WGS to analyze the relationship between mutant phenotype and genotype, discover new allelic variations, and establish a system. An efficient and accurate method for directional identification of allelic variation in rice grain type genes through mutation induction, targeted sequencing, and whole genome sequencing combined with a mixed-samples strategy, abbreviated as MTWA, was developed (Fig. 1). The innovation of this method is that the use of targeted sequencing combined with WGS can quickly screen mutants and identify mutation sites, with high detection throughput, low detection cost, and flexible target traits and sites.

Simplified steps of the MTWA process. a Wild-type (WT) seeds were irradiated with a carbon ion beam, and the M2 generation was planted and harvested. b The M2 generation mixed samples were constructed at a ratio of 8:1 for targeted sequencing detection, and the grain shape of the M2 generation seeds was measured. c Sanger sequencing was performed on individual plants in a mixed sample of potential mutations detected by targeted sequencing and compared with phenotypes to identify key mutants. d WGS of key mutants to resolve associations between mutant phenotypes and genotypes and to mine for novel allelic variants
Results
Grain Ty** Phenotype Investigation
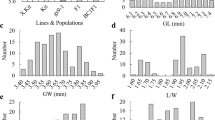
The wild-type (WT) material used in this study was Huahang No. 31 (Fig. 2a). A total of 3872 seeds of the M2 generation per plant were harvested. We measured the grain length and width of these seeds (Additional file 7; Table S4). The grain length was between 8 and 10.22 mm, the average length was 9.28 mm, and the coefficient of variation was 1.89%. The grain length of WT was 9.31 mm (Fig. 2c). The grain width was between 1.54 and 2.87 mm, with an average width of 2.02 mm, and the WT grain width was 2.03 mm, with a coefficient of variation of 0.56% (Fig. 2d, Table 1). Both the grain length and grain width conformed to a normal distribution and had a wide variation range. Compared with the WT, there were many materials with large grain type differences, indicating that there were several potential grain type mutations (Fig. 2b).

a WT Huahang 31 plant type. Scale bars = 10 cm. b Mutant granulotype and WT alignment. Scale bars = 1 cm. c Material grain length number distribution. d Material grain width number distribution
Targeted Sequencing to Screen a Mixed Pool of Potential Mutations
The target genes of targeted sequencing are GS3 (Fig. 3a) and GW5 (Fig. 3b). GS3 and GW5 are the two genes that have the greatest influence on grain length and grain width, and their mechanism has been thoroughly studied. Targeted sequencing (Fig. 3c) detected a total of 179 mutation sites were obtained (Additional file 8; Table S5), all of which were homozygous mutations, of which 110 sites were in the GS3 interval and 69 were in the GW5 interval. The total mutation frequency in the GS3 interval was calculated to be 4.05 × 10− 5, and the total mutation frequency in the GW5 interval was 9.02 × 10− 5 (total mutation frequency = mutation base number/gene fragment length). Among the 179 mutation sites, 63.57% were located in the intron region, and 30% were located in the exon region (Fig. 3d, Additional file 9; Table S6). We retained only the nonsynonymous and nonsense mutations located in the exon region that could cause amino acid changes. At the same time, reliable sites with relatively high reads were screened, and a total of 15 SNPs were obtained (Table 2), of which 14 were nonsynonymous mutations and 1 was a nonsense mutation, including 11 GS3 interval loci and 4 GW5 interval locus points (Fig. 3e), for a total of 12 mixed samples. Of the 12 mixed samples, 8 were associated with GS3 and 4 were associated with GW5. 4–101, 6–44 and 7–78 each contained two SNPs, and the remaining nine mixed samples only had one SNP.

a GS3 gene information. b GW5 genetic information. c Targeted sequencing process. d Location distribution of SNPs in the genome. e Mutation site functional type distribution
Screening of Individual Mutant Plants and Identification of Their Authenticity
To further screen out the mutant individual plants from the mixed samples, we isolated individual plants in the 12 mixed samples, which contained a total of 96 individual plant materials. The 96 individual plant materials of the fragment were subjected to Sanger sequencing and compared with the results of targeted sequencing to determine the target mutant individual plant. A total of 13 loci were consistent with the targeted sequencing results, among which the Sanger sequencing results of SNP-5 and SNP-6 were different from the targeted sequencing results (Fig. 4a), so the mutants at these two loci were excluded and targeted sequencing. The concordance rate with Sanger sequencing was 86.67%, and a total of 13 mutants were screened. The complete 15 SNP results are shown in Additional file 2: Fig. S2.
To verify the authenticity of the selected mutants, we identified the authenticity of the selected 13 single-plant materials according to the technical regulations for the identification of rice varieties (SSR marking method) issued by the Ministry of Agriculture (NY/T 1433–2014) and designed a total of 10 pairs of SSR markers (Additional file 6: Table S3). The agarose gel electrophoresis detection results of the 13 mutant individual plants were consistent with the WT, indicating that they were all true mutations (Fig. 4b).

a Sanger sequencing results; the red box is the target SNPs. The Sanger sequencing results of SNP-1 and SNP-2 were consistent with the targeted sequencing results. SNP-5 and SNP-6 were consistent with WT, and no variants were detected. b Results of agarose gel electrophoresis. The 10 SSR fragment polymorphisms of 13 mutant individual plants were consistent with WT
Phenotypic Verification and Protein Function Analysis of Individual Mutant Plants
After verification, the corresponding grain type and phenotype data of the real variant individual plants were found according to the number. Only 6 of the 13 individual plants showed significant changes in grain type, and the grain types of the remaining 7 individual plants were the same as those of the control. There was no significant difference in the ratios between samples (Table 3). According to the screening results of targeted sequencing, 13 SNPs are nonsynonymous and nonsense mutations, which theoretically lead to amino acid changes, while some SNPs do not cause significant changes in phenotype, presumably not changing the function of a protein or structural or other genetic mutations.
We screened a total of 9 grain length mutants, of which 2 grain lengths were significantly longer and 2 grain lengths were significantly shorter than WT grains (Fig. 5a). Nine SNPs related to GS3 were identified, including 8 nonsynonymous mutations and 1 nonsense mutation (GS3-2), in which GS3-1 was located in exon 1 and GS3-2 was located in exon 2. The remaining seven mutations were located in exon 5 (Fig. 5b). The mutation position of GS3-1 is relatively advanced, and it is not located in the functional structural region and has no effect on the structure and function of the protein, so the grain length does not change significantly (Fig. 5b). GS3-2 is located in the OSR domain, the 55th amino acid is mutated to a stop codon, the OSR domain is deleted, and the protein structure and function are severely affected (Fig. 5d), resulting in a significant increase in grain length. Both GS3-3 and GS3-4 are located in the TNFR domain of Cys-rich mutants, and the grain length of GS3-3 is significantly reduced. Protein structural analysis showed that the mutation of amino acid No. 135 leads to two additional β sheets in the secondary structure of the protein. and presumably resulted in impaired TNFR domain function (Fig. 5d), whereas the GS3-4 grain length was not significantly altered. GS3-5, GS3-6, GS3-7, GS3-8 and GS3-9 are all located in the Cys-rich VWFC domain, among which only the grain length of GS3-5 is significantly reduced, and the mutation of GS3-5 may lead to the impaired structure of the VWFC domain function, but there is no significant difference in protein structure compared with WT plants (Additional file 3: Fig. S3). The GS3-6, GS3-8 and GS3-9 phenotypic results were similar to those of GS3-4; functional domain amino acid point mutation occurred, but the phenotype did not change significantly. It was speculated that the mutation of these 4 amino acids may not affect the function of the protein or that other gene mutations have an impact on the phenotype; however, the grain length of GS3-7. In contrast, the functional site analysis of its protein showed that the mutation of amino acid 183 of GS3-7 was located in the ligand binding site of the protein (Fig. 5c). After the mutation, the function of the protein was affected, so the particle shape changed. However, GS3-4, GS3-5, GS3-6, GS3-8 and GS3-9 showed no significant difference when compared to the WT in terms of protein structure and function (Additional file 3: Fig. S3).

a Grain length and WT alignment of nine GS3 mutants. Scale bars = 1 cm. b Mutant distribution on gene and protein structure. c GS3 protein functional site (red part). d Protein structures of GS3 mutants (differential structures in red)
We screened 4 mutants with grain width, of which 1 grain width increased, 1 grain width decreased, and the remaining two grain widths had no significant changes (Fig. 6a). The four identified GW5-related SNPs were all nonsynonymous mutations, of which GW5-4 was located in exon 1, and the other three were located in exon 2 (Fig. 6b). GW5-1, GW5-2 and GW5-3 are all located in the calmodulin-binding domain; the difference is that only the granule width of GW5-1 is significantly wider than that of other mutants, and the other two mutants have no obvious change in phenotype. We predicted the protein structure and function of mutants and found that the protein structures of GW5-1 and GW5-2 were more similar to each other than to the WT, while the protein structure of GW5-3 had no obvious change (Fig. 6c). The phenotypes of GW5-1 and GW5-2 of the same domain differ, presumably due to interference from other genes. The GW5-4 mutation position is relatively forward, not located in the functional structural region, and has no effect on the structure and function of the protein (Fig. 6b), but the grain width is significantly narrowed, and the protein structure is relatively concentrated (Fig. 6c).

a Grain width and WT alignment of four GW5 mutants. Scale bars = 1 cm. b Mutant distribution on gene and protein structure. c Protein structures of GW5 mutants (differential structures in red)
Whole Genome Sequencing of Key Mutants Identifies New SNPs Affecting the GS3 Mutation Effect.
To explore the reasons for the contradiction between genotype mutation and phenotype mutation and to clarify whether allelic variation in other grain length-related genes had an impact on the phenotype, GS3-M1, GS3-M2 and WT sample were subjected to WGS. A total of 2,084,534 SNPs and 336,039 InDels were obtained by sequencing GS3-M1, and 2,116,343 SNPs and 341,777 InDels were obtained from GS3-M2. There were 189473 different loci in the two samples after comparison (Additional file 10; Table S7). After screening, three new allelic variants related to grain length were finally obtained (Table 4) (Fig. 7a). GS3-G1 is located in the second exon of OsNST1, which mutates serine No. 65 to threonine. At present, there are few reports on this gene, and its protein structure cannot be predicted. Mutants exhibit reduced cell wall cellulose content and structural changes, resulting in reduced mechanical strength and abnormal plant development, such as dwarf plants and smaller seed size (Song et al. 2011). GS3-G2 is a variant located in the first exon of OsMAPK6 that mutates the aspartic acid at No. 131 to arginine, which affects only one of its functional domains. Inhibition of OsMPK6 expression can make rice panicles denser and grains smaller, and mutation of this gene can significantly reduce grain length, grain width and thousand-grain weight (Guo et al. 2018). GS3-G3 is located in the second exon of RAE2, resulting in a frameshift insertion mutation at amino acid 99 and impaired function of the cysteine-rich region of the encoded protein EPFL1. The number of kernels decreased, the kernels became longer, and the proportion of awned kernels increased (** et al. 2016) (Fig. 7b).

a Whole-genome mutation screening process. b Positions of the three new SNPs on their corresponding genes
Discussion
Reliability of MTWA to Identify Mutants and Allelic Variants
Screening mutants and identifying allelic variants are important foundations for innovative germplasm materials and functional genomics research (Guo et al. 2006). To efficiently utilize germplasm resources, the most fundamental way is to excavate new alleles and purposefully aggregate or transfer them in conventional breeding or molecular breeding and then combine them with molecular design to achieve the purpose of improving breeding efficiency. In this study, the seeds of Huahang 31 were irradiated with 12C6+, and the M2 generation population containing 3872 individual plants was obtained. The variation coefficient of grain length was 1.89%, and the variation coefficient of grain width was 0.56%, indicating that there are many potential mutations in grain type. body. In this study, a new method for allelic variation and mutant identification, MTWA, was proposed, which used targeted sequencing technology to initially identify mixed sample materials. After screening, a total of 15 SNPs and 12 mixed sample materials were obtained, and Sanger sequencing was performed. The key mutants in the mixed samples were screened out and verified for authenticity. The consensus rate between targeted sequencing and Sanger sequencing was 86.67%, proving the feasibility of the method. A total of 13 mutants and 15 SNPs were screened out. Analysis of the phenotype of the mutant individual plant, the protein functional structural analysis of the mutation site, and WGS of the key mutants were conducted. Mining new grain length-related allelic variations, analyzing the connection between genotype mutations and phenotype mutations, and establishing a set of systematic, efficient and accurate new methods for allelic variation identification were also conducted. At the same time, a batch of mutation sites and mutant materials with breeding value were screened and identified.
Advantages of MTWA
Traditional forward genetic identification methods usually require the construction of complex map** populations and linkage maps and gene linkage analysis using genetic markers (Zhang et al. 2012; Serquen et al. 1997; Fazio et al. 2003) but can only map mutation sites to a large range of chromosomal regions (Hazen 2005), and fine map** of mutant genes is expensive and time consuming (Schneeberger et al. 2009; Abe et al. 2012). MTWA can be utilized to analyze the M2 generation without the need to construct a complex genetic population, which greatly shortens the detection time. This technique is also a directional mutant identification method. TILLING technology uses CEL I enzyme to digest PCR amplification products and detects and selects mutants by capillary electrophoresis (Yan et al. 2014), but the process is relatively complicated; however, the TILLING technique can detect gene fragments. There are certain length requirements; usually, the length of the target gene fragment is less than 1.5 kb, and the high-resolution melting curve (HRM) detection region is only 150–500 bp (Sikora et al. 2011). On the other hand, MTWA directly performs second-generation sequencing on the target fragment to determine the mutation sites. The MTWA process is relatively simple, and the sequencing results are more accurate than those of the TILLING method. At the same time, multiple mutation sites can be detected in a single amplicon, and there is no restriction on the fragment length. The detection efficiency is higher and has wider applicability than the TILLING method. Compared to MutMap and its derived methods, mutants in MTWA do not require backcrossing of parental lines, thus greatly reducing time and effort (Allen et al. 2013; Abe et al. 2012); if the genome of the target crop is large and complex, MutMap will have the problems of high sequencing costs, large datasets, and difficult comparison and analysis, especially in allopolyploid species with high genome heterozygosity, and highly homologous sequences and subtypes of genomes could also be detected (Li et al. All data generated or analyzed during this study are included in this published article and its supplementary information files. Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiruet M (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30(2):174–178. https://doi.org/10.1038/nbt.2095 Allen RS, Nakasugi K, Doran RL, Millar AA, Waterhouse PM (2013) Facile mutant identification via a single parental backcross method and application of whole genome sequencing based map** pipelines. Front Plant Sci 4:362. https://doi.org/10.3389/fpls.2013.00362 Anai T (2012) Potential of a mutant-based reverse genetic approach for functional genomics and molecular breeding in soybean. Breed Sci 61(5):462–467. https://doi.org/10.1270/jsbbs.61.462 Boualem A, Fleurier S, Troadec C, Audigier P, Kumar APK, Chatterjee M, Alsadon AA, Sadder MT, Wahb-Allah MA, Al-Doss AA, Bendahmane A (2014) Development of a Cucumis sativus TILLING platform for forward and reverse genetics. PLoS ONE 9(5):e97963. https://doi.org/10.1371/journal.pone.0097963 Chen A, Dubcovsky J (2012) Wheat tilling mutants show that the vernalization gene VRN1 down-regulates the flowering repressor VRN2 in leaves but is not essential for flowering. PLoS Genet 8(12):e1003134. https://doi.org/10.1371/journal.pgen.1003134 Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17):i884–i890. https://doi.org/10.1093/bioinformatics/bty560 Consortium T (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345(6194):1251788. https://doi.org/10.1126/science.1251788 Du H, Yang J, Chen B, Zhang X, Wen C (2019) Target sequencing reveals genetic diversity, population structure, core-SNP markers, and fruit shape-associated loci in pepper varieties. BMC Plant Biol 19(1):578. https://doi.org/10.1186/s12870-019-2122-2 Fan C, **ng Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3,a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theoretical & Applied Genetics 112(6):1164–1171. https://doi.org/10.1007/s00122-006-0218-1 Fazio G, Staub JE, Stevens MR (2003) Genetic map** and QTL analysis of horticultural traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Theoretical & Applied Genetics 107(5):864–874. https://doi.org/10.1007/s00122-003-1277-1 Guo L, Chu C, Qian Q (2006) Rice mutants and functional genomics. Chin Bull Bot 23(1):1–13. https://doi.org/10.3969/j.issn.1674-3466.2006.01.001 Guo T, Chen K, Dong NQ, Shi CL, Ye WW, Gao JP (2018) Grain size and number1 negatively regulates the OsMKKK10-OsMKK4-OsMPK6 cascade to coordinate the trade-off between grain number per panicle and grain size in rice. Plant Cell 30(4):871–888. https://doi.org/10.1105/tpc.17.00959 Guo Z, Wang H, Tao J, Ren Y, Xu C, Wu K, Zou C, Zhang J, Xu Y (2019) Development of multiple SNP marker panels affordable to breeders through genoty** by target sequencing (GBTS) in maize. Mol Breeding 39(3):37. https://doi.org/10.1007/s11032-019-0940-4 Hase Y, Yoshihara R, Nozawa S, Narumi I (2012) Mutagenic effects of carbon ions near the range end in plants. Mutat Research/fundamental Mol Mech Mutagen 731(1–2):41–47. https://doi.org/10.1016/j.mrfmmm.2011.10.004 Hazen PS (2005) Rapid array map** of circadian clock and developmental mutations in arabidopsis. Plant Physiol 138(2):990–997. https://doi.org/10.1104/pp.105.061408 Henikoff S, Comai L (2003) Single-nucleotide mutations for plant functional genomics. Annu Rev Plant Biol 54(1):375–401. https://doi.org/10.1146/annurev.arplant.54.031902.135009 ** J, Hua L, Zhu Z, Tan L, Zhao X, Zhang W, Liu F, Fu Y, Cai H, Sun X (2016) GAD1 encodes a secreted peptide that regulates grain number, grain length and awn development in rice domestication. Plant Cell 28(10):2453–2463. https://doi.org/10.1105/tpc.16.00379 **g Y, Gao F, Liu L, Su X, Ren M, Lu X, Lyu J, ** W, Hu Y, Ren J, Ren G (2021) Phenotype and genome variation analysis of ion-beam induced mutant rice JD-1. J Plant Genetic Resour 22(05):1322–1333. https://doi.org/10.1016/j.mrfmmm.2021.111757 Li F, Fan G, Lu C, **ao G, Zou C, Kohel RJ, Xu X (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33(5):524–530. https://doi.org/10.1038/nbt.3208 Li F, Shimizu A, Nishio T, Tsutsumi N, Kato H (2019) Comparison and characterization of mutations induced by gamma-ray and carbon-ion irradiation in rice (Oryza sativa L.) using whole-genome resequencing. G3-Genes Genomes Genetics 9(11):3743–3751. https://doi.org/10.1534/g3.119.400555 Li X, Zheng H, Wu W, Liu H, Zhao H (2020) QTL map** and candidate gene analysis for alkali tolerance in japonica rice at the bud stage based on linkage map** and genome-wide association study. Rice 13(1):1–11. https://doi.org/10.1186/s12284-020-00412-5 Ling HQ, Zhao S, Liu D, Wang J, Sun H, Al E (2013) Draft genome of the wheat A-genome progenitor triticum urartu. Sci Found China 0237. https://doi.org/10.1038/nature11997 Liu J, Chen J, Zheng X, Wu F, Lin Q, Heng Y, Tian P, Cheng ZJ, Yu X, Zhou K (2017) GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat Plants 3(5):951–957. https://doi.org/10.1038/nplants.2017.43 Lu Q, Hong Y, Li S, Liu H, Li H, Zhang J, Lan H, Liu H, Li X, Wen S (2019) Genome-wide identification of microsatellite markers from cultivated peanut (Arachis hypogaea L.). BMC Genomics 20(1):799. https://doi.org/10.1186/s12864-019-6148-5 Mao H, Sun S, Yao J, Wang C, Zhang Q (2010) Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci 107(45):19579–19584. https://doi.org/10.1073/pnas.1014419107 McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110 Michael A, Jane R, Thomas L, Raphaël F, Françoise A, Cyril P, Nacer M, Sophie G, Erik K, Célia M (2018) Linking the international wheat genome sequencing consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol 19(1):1–10. https://doi.org/10.1186/s13059-018-1491-4 Minoia S, Petrozza A, D"Onofrio O, Piron F, Mosca G, Sozio G, Cellini F, Bendahmane A, Carriero F (2010) A new mutant genetic resource for tomato crop improvement by TILLING technology. BMC Res Notes 3(1):69. https://doi.org/10.1186/1756-0500-3-69 Miura K, Ikeda M, Matsubara A, Song XJ, Ito M, Asano K, Matsuoka M, Kitano H, Ashikari M (2010) OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat Genet 42(6):545–549. https://doi.org/10.1038/ng.592 Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4325. https://doi.org/10.1093/nar/8.19.4321 NY/T 1433—Protocol for identification of rice varieties-SSR marker method Ochiai K, Shimizu A, Okumoto Y, Fujiwara T, Matoh T (2011) Suppression of a NAC-Like transcription factor gene improves boron-toxicity tolerance in rice. Plant Physiol 156(3):1457–1463. https://doi.org/10.1104/pp.110.171470 Okasa AM, Sjahril R, Riadi M, Mahendradatta M, Sato T, Toriyama K, Ishii K, Hayashi Y, Abe T (2021) Correlation and path coefficient analysis of grain yield and its components in toraja land-race aromatic rice mutants induced by heavy ion beam. Asian J Plant Sci 20(3):406–413. https://doi.org/10.3923/ajps.2021.406.413 Oono Y, Ichida H, Morita R, Nozawa S, Hase Y (2020) Genome sequencing of ion-beam-induced mutants facilitates detection of candidate genes responsible for phenotypes of mutants in rice. Mutat Research/Fundamental Mol Mech Mutagen 821:111691. https://doi.org/10.1101/666677 Qu Y, LI W, Zhou L, Wang C, Dong C, Yu L, Liu Q, He J (2007) Research and application of mutagenic effects in plants irradiated by heavy ion beams. Nuclear Phys Rev 24(4):294–298. https://doi.org/10.3969/j.issn.1007-4627.2007.04.010 Rym F, Hiroki T, Muluneh T, Akira A, Satoshi N, Hiroki Y, Shailendra S, Shiveta S, Hiroyuki K, Hideo M (2017) MutMap+: genetic map** and mutant identification without crossing in rice. PLoS ONE 8(7):e68529. https://doi.org/10.1371/journal.pone.0068529 Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL, Jrgensen JE, Weigel D, Andersen SU (2009) SHOREmap: simultaneous map** and mutation identification by deep sequencing. Nat Methods 6(8):550–551. https://doi.org/10.1038/nmeth0809-550 Serquen FC, Bacher J, Staub JE (1997) Map** and QTL analysis of horticultural traits in a narrow cross in cucumber (Cucumis sativus L.) using random-amplified polymorphic DNA markers. Mol Breeding 3(4):257–268. https://doi.org/10.1023/A:1009689002015 Shen Y, Wang J, Shaw RK, Yu H, Sheng X, Zhao Z, Gu H (2021) Development of GBTS and KASP panels for genetic diversity, population structure, and fingerprinting of a large collection of broccoli (Brassica oleracea L. var. italica) in China. Front Plant Sci 12:655254. https://doi.org/10.3389/fpls.2021.655254 Sikora P, Chawade A, Larsson M, Olsson J, Olsson O (2011) Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int J Plant Genomics 2011:314829. https://doi.org/10.1155/2011/314829 Sjahril R, Trisnawaty AR, Riadi M, Rafiuddin R, Sato T, Toriyama K, Hayashi Y, Abe T (2020) Selection of early maturing and high yielding mutants of toraja local red rice grown from M2-M3 population after ion beam irradiation. Bogor Agricultural University 27(2):166–173. https://doi.org/10.4308/HJB.27.2.166 Song X, Zhang B, Zhou Y (2011) Golgi-localized UDP-glucose transporter is required for cell wall integrity in rice. Plant Signal Behav 6(8):1097–1100. https://doi.org/10.4161/psb.6.8.16379 Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C (2013a) QTL-seq: rapid map** of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74(1):174–183. https://doi.org/10.1111/tpj.12105 Takagi H, Tamiru M, Abe A, Yoshida K, Uemura A, Yaegashi H, Obara T, Oikawa K, Utsushi H, Kanzaki E (2015) MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol 33(5):445–449. https://doi.org/10.1038/nbt.3188 Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C, Utsushi H, Natsume S, Kanzaki H, Matsumura H, Saitoh H, Yoshida K, Cano LM, Kamoun S, Terauchi R (2013b) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies he trice blast resistance gene Pii. New Phytol 200(1):276–283. https://doi.org/10.1111/nph.12369 Yan Z, Guo H, Xu R, Liu L (2014) Development of TILLING technology and its application in different plants. J Nuclear Agricul Tural Sci 28(02):224–233. https://doi.org/10.11869/j.issn.100-8551.2014.02.0224 Yang G, Luo W, Zhang J, Yan X, Du Y, Zhou L, Guo T (2019a) Genome-wide comparisons of mutations induced by carbon-ion beam and gamma-rays irradiation in rice via resequencing multiple mutants. Front Plant Sci 10:1514. https://doi.org/10.3389/fpls.2019.01514 Yang J, Zhang J, Han R, Zhang F, Mao A, Luo J, Dong B, Liu H, Tang H, Zhang J (2019b) Target SSR-seq: a novel ssr genoty** technology associate with perfect SSRs in genetic analysis of cucumber varieties. Front Plant Sci 10:531. https://doi.org/10.3389/fpls.2019.00531 Zhang B, Liu X, Qian Q, Liu L, Dong G, **ong G, Zeng D, Zhou Y (2011) Golgi nucleotide sugar transporter modulates cell wall biosynthesis and plant growth in rice. Proc Natl Acad Sci USA 108(12):5110–5115. https://doi.org/10.2307/41125277 Zhang C, Zhu J, Chen S, Liu Q (2019) Wxlv, the ancestral allele of rice Waxy gene. Mol Plant 12(8):1157–1166. https://doi.org/10.3390/proceedings2019036140 Zhang WW, Pan JS, He HL, Zhang C, Li Z, Zhao JL, Yuan XJ, Zhu LH, Huang SW, Cai R (2012) Construction of a high density integrated genetic map for cucumber (Cucumis sativus L.). Theor Appl Genet 124(2):249–259. https://doi.org/10.1007/s00122-011-1701-x Zhang X, Yang F, Ma HY, Li JP (2022) Evaluation of the saline-alkaline tolerance of rice (Oryza sativa L.) mutants induced by heavy-ion beam mutagenesis. Biology 11(1):126. https://doi.org/10.3390/biology11010126 Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J (2011) Genome-wide association map** reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2(1):467. https://doi.org/10.1038/ncomms1467 Zheng Y, Li S, Huang J, Fu H, Zhou L, Furusawa Y, Shu Q (2021) Identification and characterization of inheritable structural variations induced by ion beam radiations in rice. Mutat Research/Fundamental Mol Mech Mutagen 823:111757. https://doi.org/10.1016/j.mrfmmm.2021.111757 The authors would thank American Journal Experts for their valuable language service. This work was supported by the Special Rural Revitalization Funds of Guangdong Province (2021KJ382) and the Research and Development Plan for Key Areas in Guangdong Province (No. 2018B020206002). KS and TG designed the experiment and wrote the manuscript. KS, DDL, AYX, QW and SSJ conducted the experiments and performed data analysis. HZ, JFW, GLY, DHZ, CHH, HW and ZQC drafted proposals and corrected the manuscript. All authors read and approved the final manuscript. This study complied with the ethical standards of China where this research was performed. Not applicable. The authors declare no conflict of interest. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. VAF calculation method, green bases are mutation bases, red bases are reference genome bases, and the ratio of green bases to all bases is the VAF. The Sanger sequencing results of the mutation sites of the 15 mutant individual plants, in which the results of SNP-5 and SNP-6 are different from the targeted sequencing results, these two mutant individual plants are excluded, and the rest are the same as the targeted sequencing results, which are true variation. Protein structures of 9 GS3 mutants, the red part is the difference from WT. Targeted sequencing primer information. Primers for amplifying fragments of the target site. Primers for authenticity verification of mutant individuals. Grain type datas. Mutation mixed sample information screened by targeted sequencing. Mutation site information obtained by targeted sequencing. Mutation site information screened by whole genome sequencing. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Sun, K., Li, D., **a, A. et al. Targeted Identification of Rice Grain-Associated Gene Allelic Variation Through Mutation Induction, Targeted Sequencing, and Whole Genome Sequencing Combined with a Mixed-Samples Strategy.
Rice 15, 57 (2022). https://doi.org/10.1186/s12284-022-00603-2 Received: Accepted: Published: DOI: https://doi.org/10.1186/s12284-022-00603-2Availability of data and materials
References
Acknowledgements
Funding
Author information
Authors and Affiliations
Contributions
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Additional information
Publisher’s Note
Supplementary Information
Additional file 1: Fig. S1.
Additional file 2: Fig. S2.
Additional file 3: Fig. S3.
Additional file 4: Table S1.
Additional file 5: Table S2.
Additional file 6: Table S3.
Additional file 7: Table S4.
Additional file 8: Table S5.
Additional file 9: Table S6.
Additional file 10: Table S7.
Rights and permissions
About this article
Cite this article
Keywords
