
Abstract
The dysregulation of apoptosis contributes in a variety of ways to the malignant phenotype. It is increasingly recognized that the alteration of pro-apoptotic and anti-apoptotic molecules determines not only escape from mechanisms that control cell cycle and DNA damage, but also endows the cancer cells with the capacity to survive in the presence of a metabolically adverse milieu, to resist the attack of the immune system, to locally invade and survive despite a lack of tissue anchorage, and to evade the otherwise lethal insults induced by drugs and radiotherapy. A multitude of apoptosis mediators has been identified in the past decade, and the roles of several of them in breast cancer have been delineated by studying the clinical correlates of pathologically documented abnormalities. Using this information, attempts are being made to correct the fundamental anomalies at the genetic level. Fundamental to this end are the design of more efficient and selective gene transfer systems, and the employment of complex interventions that are tailored to breast cancer and that are aimed concomitantly towards different components of the redundant regulatory pathways. The combination of such genetic modifications is most likely to be effective when combined with conventional treatments, thus robustly activating several pro-apoptotic pathways.
Similar content being viewed by others


Introduction
The highly orchestrated form of cell death known as apoptosis goes awry to some extent in most cancers. Increasingly, a general theme in cancer pathophysiology is the development of a defect in the function of pro-apoptotic molecules, such as p53, that commonly prepare the cell for apoptosis whenever cell proliferation or DNA damage is induced; lack of these molecules therefore deprives the cell of a critical safety mechanism [1]. Alternatively, a functional excess of anti-apoptotic molecules, such as Bcl-2, may also occur in tumors. In each case, the result is an imbalance that favors the inappropriate survival of tumor cells. The mechanisms involved and their components are attractive therapeutic targets because the tumor cell is totally dependent on them for its survival, and appears typically to have a higher sensitivity to the induction of apoptosis than normal tissues [1,2]. In addition, restoring or enhancing the capacity to undergo apoptosis may, in some cases, be a crucial event that renders tumors sensitive to classical anticancer agents, such as those used in chemotherapy [3,4**] and radiotherapy [5,6]. The present review discusses the most common genetic alterations that alter apoptosis regulation in breast cancer, and the initial gene-based approaches that have been explored to overcome them with therapeutic intent.
Dysregulated apoptosis contributes to cancer
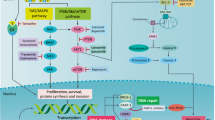
There is a multitude of critical steps during the pathogenesis of cancer in which avoidance of programmed cell death assures the progression, and maintenance, of the malignant phenotype (Fig. 1) [7,8*,9,10]. Early during tumor progression, defects in apoptosis allow the survival of the cancer cell despite the existence of DNA damage and cell-cycle dysregulation. This severe breaking of a basic DNA housekee** action contributes to the genetic instability that characterizes cancer, and thus initiates and sustains a spiral of further and further genetic aberrations that endow tumor cells with extraordinary capacities and adaptability. In fact, a reduction in apoptosis has been observed in patients with both carcinomas and 'normal' epithelium in the breast that is associated with fibrocystic changes. This is in contrast with breasts with benign fibroadenomas, which have normal levels of apoptosis and are not associated with malignant transformation [11*]. Further evidence has been obtained in transgenic mice that express the oncogenic simian virus 40 (SV40) large T antigen. These animals show a dramatic increase in apoptosis during the development of preneoplastic mammary lesions, which is associated with a significant elevation in expression of the pro-apoptotic Bax. In double-transgenic mice that are engineered to additionally carry a mutated bax, a marked reduction in apoptosis occurs in the preneoplastic lesions and the animals show a subsequent increase in the number, size, and rate of growth of breast tumors [12*]. Thus, disruption of apoptosis mediators clearly contributes to mammary tumor progression.

Dysregulation of apoptosis contributes to the pathophysiology ofcancer. A variety of defects in the apoptotic machinery contribute to avoidanceof apoptosis by tumor cells throughout the entire carcinogenic process. Theenhanced capacity of tumor cells to survive allows them to overcome (in analogyto decathlon athletes) numerous challenges encountered, not only in the primarytumor location, but also during their vascular distribution and at theirmultiple final destinations. The universal dependence of tumor cells onmechanisms to avoid apoptosis suggests a 'window of homogeneity'that could be exploited therapeutically. CTL, cytotoxic T lymphocyte.
As the tumor mass grows, the demands for oxygen, basic nutrients, and growth factors are increasingly unmet. This imbalance, which would otherwise induce cell death, is not translated into effective death signals by a disturbance in the apoptotic machinery, however. Furthermore, the accumulating mutations of the tumor cell give rise to new cell surface mutated proteins, or epitopes. These potential tumor antigens, however, do not usually generate a strong immune response, or the response is not efficacious. Such 'sheltering' from cytotoxic T lymphocytes (CTLs) and other immune effectors has been associated with a variety of changes in the control of apoptosis that effectively protect the cancer cell from otherwise lethal insults mediated by CTLs and other immune mediators [13].
Later in the natural history of malignant tumors, metastases eventually develop, which determines in most cases an ominous change in the prognosis of the disease. For this event to occur the cell needs to acquire the capacity to disseminate and survive despite its lacking tissue anchorage, which normally would induce a type of apoptosis called 'anoikis'. Recently, the involvement of cellular receptors of death signals in anoikis has been described [14**], and the disturbance of these receptors and their related transductional apparatus in cancer cells has been suggested. Lastly, the tumor regression induced by chemotherapy, radiotherapy, and hormone therapy depends to a large extent on the induction of apoptosis [7,15,16,17]. In breast cancer, the administration of chemotherapy immediately before surgery has allowed analysis of the induction of apoptosis in the remaining tumor within the resected specimen. Interestingly, a positive correlation has been found between the apoptotic index, the clinical response, and patient survival [18**]. Tumor cells frequently emerge, however, that resist the apoptosis-inducing effects of those maneuvers, thus essentially esca** from current therapeutic interventions [19,20,21]. On aggregate, the consequences of dysregulation of apoptosis are pervasive throughout the natural history of cancer, and have been established as a universal component of the malignant phenotype.
Mechanisms of apoptosis avoidance in breast cancer
In cancer, cellular proliferation goes on unchecked and cellular death does not occur to the extent that it should, despite several otherwise potent death stimuli being conspicuously present. These two aspects of tumor pathophysiology have been analyzed in breast cancer, and consistently found to be disturbed [22,23,24]. In fact, expression levels of several cell growth regulators and pro-apoptotic and anti-apoptotic proteins are usually perturbed in breast cancer, and their alteration has been associated with prognosis and with the response to conventional anticancer treatments (see below).
Growth factors and their receptors
Many growth factors and their receptors influence the growth and proliferation of breast cancer cells [25]. Typically, normal versions of human epidermal growth factor receptor-related gene (HER)-2/neu (c-erbB-2), epidermal growth factor receptor (or c-erbB-1), and insulin-like growth factor (IGF)-1 receptor are overexpressed. Interestingly, the aberrant control of cell proliferation by growth factor receptors is mechanistically related to an inhibition of apoptosis. For instance, overexpression of HER-2 upregulates anti-apoptotic bcl-2 and bcl-xL, and thus inhibits in vitro tamoxifen-induced apoptosis [26]. A similar effect of HER-2 has been found on taxol-induced apoptosis. As another example, IGF-1 protects breast cancer cells from apoptosis that is induced by chemotherapeutic drugs [27]. Thus, mechanisms known to alter tumor cell proliferation may also directly contribute to the avoidance of apoptosis in breast cancer cells. The relevant molecular pathology, and the potential for modulating these molecules in the context of gene therapy, has been reviewed elsewhere [28,29].
Genes that regulate apoptosis
In addition to factors that are involved in controlling cell proliferation, abnormalities have been identified in breast cancers in many genes that regulate the apoptotic cascade, including p53, bcl-2, bax, c-myc, p21 WAF/CIP1, and many others (Table 1). As a consequence, tumor cells express several proteins that render them resistant to apoptosis. Blocking cell death promotes neoplastic transformation [10].
p53
Mutations in the p53 gene are a common molecularabnormality in breast cancer [30,31]. A consequence of the lack of normal function ofp53 may be the failure to induce apoptosis in cells with damaged DNA[32], and it can also possibly impair a full apoptoticresponse to the administration of hormonal or chemotherapeutic interventions.Furthermore, it can contribute to genomic instability [33], and thus increase the probability of appearance ofadditional mutations that are advantageous for survival of the tumor cell. p53stops the cell cycle and induces apoptosis through stimulation ofp21WAF/CIP1, an inhibitor of cyclin-dependent kinases. In effect,experimental overexpression of p21WAF/CIP1 in human breast cancercell lines suppresses growth, and induces apoptosis [34].
Bcl-2 family
The genes of the bcl-2 family have emerged as keyregulators of apoptosis, and appear to be dysregulated in a number of tumors,including breast cancers [23,35,36]. Several members of the Bcl-2family, including bcl-2, Bcl-XL, Mcl-1, and A1/Bfl-1, suppressapoptosis; whereas others, including Bax, Bak, Bok/Mtd, Bad, Bik, Bid, Bim/Bod,and HrK, induce apoptosis. The extent of apoptosis is inversely associated withBcl-2 expression in pre-malignant and malignant breast lesions [37,38,39,40]. Paradoxically, Bcl-2 expression correlates with favorableclinicopathologic features, as well as with improved disease-free and overallsurvival [41,42,43,44,45,46,47]. Furthermore, patients withelevated Bcl-2 levels appear to derive the greatest benefit from endocrinetherapy [48,49,50]. Only in a subset of well differentiated and progesteronereceptor-positive tumors has Bcl-2 been reported to enhance disease progression[35]. As an explanation for the apparent paradox ofdecreasing levels of anti-apoptotic Bcl-2 levels with increasing tumor grade,it has been proposed that Bcl-2 has an early role within the tumor by rescuingcells with otherwise lethal mutations. After additional oncogene activation,some cells would acquire additional ways to protect themselves againstapoptosis [51]. At that point, loss of Bcl-2 mightconfer a growth advantage. In fact, Bcl-2 is known to restrain cellproliferation [10]. Thus, expression of Bcl-2 wouldchange from high levels in early or low-grade tumors, characterized by lowapoptotic indices, to low levels in advanced or high-grade tumors,characterized by high apoptotic indices.
Bax
The levels of expression of Bax or the Bax:Bcl-2 ratio, asdetermined by immunostaining, directly correlate in breast tumors with longerpatient survival [46,47] and/orbetter response to therapy in some [52] but not allstudies [53]. In this regard, bax is considereda tumor suppressor gene, and is itself a direct transcriptional target ofp53. Interestingly, haploid loss of bax leads to acceleratedmammary tumor development in SV40 large T antigen double trans-genic mice[12*], which suggest that the protective effect ofbax is dose-dependent. In addition, successful induction of apoptosisby chemotherapy and radiation is associated with augmented expression of Bax[54]. Furthermore, induced overexpression of Bax renderstumor cells more sensitive to drugs [55], whereasablation of Bax reduces drug-induced apoptosis [56].Mechanistically, Bax promotes cell death by directly binding and antagonizingpro-survival Bcl-2 [57], and it also induces apoptosisby itself through its direct channel-forming activity in mitochondria andactivation of caspase pathway [66]. In addition to anti-apoptotic Bcl-2 and analogs, other relevant proteins are the inhibitor of apoptosis protein (IAP) family and the heat shock proteins (HSPs). The IAP family members directly bind and inhibit certain caspases [67,68]. Given the central role of caspases in apoptosis, and the role of apoptosis in the cytotoxicity of chemotherapy and radio-therapy, it follows that IAPs protect tumor cells from several drugs and other therapies that induce apoptosis [68]. Information about the expression of most IAP members in human tumors, including breast cancer, is scarce. One exception is the IAP named survivin, which is known to be highly overexpressed in breast and most other cancers [69**,70], thus supporting the concept that increased activity of IAPs contributes to breast tumorigenesis. Interestingly, survivin may counteract a default induction of apoptosis in G2/M phase [71]. Its overexpression may therefore overcome an apoptosis-related cell cycle checkpoint and favor aberrant progression of transformed cells through mitosis. Of note, transcriptional activation of IAPs is dependent on nuclear factor-κB (NF-κB). In turn, IAPs can activate NF-κB and thus create a positive feedback loop, enhancing survival signals. This loop may in part explain the potent anti-apoptotic activity of NF-κB [66], and also suggests a possible target for therapeutic intervention.
Heat shock proteins
HSPs are involved in cellular resistance to stress. They are among the most conserved proteins in phylogeny, indicating their central role in supporting cell survival not only after heat shock, but also most other apoptotic stimuli [66]. Clinicopathologic studies [72,73,74,75] have shown that expression of HSP-70 occurs in high-grade tumors and correlates in breast cancer with shorter disease-free survival, increased cell proliferation, and poor differentiation, as well as lymph node involvement. In addition, expression of HSP-70 inversely correlates with the response to combination chemotherapy, radiotherapy, and hyperthermia, whereas no correlation has been found with the response to tamoxifen [75,76,77]. Interestingly, HSP-70 can rescue cells from apoptosis induced by TNF, even after the activation of effector caspases, which indicates that it has a markedly downstream point of action in the apoptosis pathway and is therefore a particularly attractive therapeutic target.
Gene therapeutics for modulation of apoptosis
The consequences of dysregulation of apoptosis on tumor pathophysiology are extremely diverse (see above and Fig. 1). The core machinery of cell death, however, seems to be formed by a discreet number of protein families (Fig. 2). Modulation of a limited number of targets by delivery of genes that encode apoptosis-related proteins would therefore have multiple beneficial effects on the malignant phenotype. Not only would tumor cell proliferation be properly balanced by increased cell death, but also barriers to the immune response would fall, tumor invasiveness would be crippled, and sensitivity to the cytotoxicity induced by drugs and radiation would be restored. Importantly, the need that tumor cells have for mechanisms that allow them to avoid apoptosis is universal, and shared by all tumor localizations in the body. This avoidance represents a 'window of homogeneity' that can be targeted in the context of the formidable tumor heterogeneity. With the increasing recognition of the molecular basis of the apoptotic pathway [1,60,78,79,80], and the description of several of its components acting as oncogenes or tumor suppressor genes, gene therapy has thus emerged as a rational strategy for the modulation of apoptosis [20,81,82,83,84]. The following is a brief summary of approaches that have already been clinically explored in the context of breast cancer. A discussion of the requirements for successful exploitation of the apoptotic machinery using gene transfer and some of the solutions being developed is then provided.
Regulation of cell death. Apoptosis involves a sensor that detectspro-apoptotic stimuli, a signal transduction network, and execution machinery.Despite the complexity of its regulation, execution of programmed cell death iseffected by the well-defined family of caspases. Upstream, at least one familyof proteins exist at each level of response to pro-apoptotic stimuli that isable to block a deadly signal, including the heat shock proteins (HSPs), theanti-apoptotic death effector domain proteins (ADEDs), several members of theBcl-2 family, the inhibitors of apoptosis proteins (IAPs), and the nuclearfactor-κB (NF-κB) family of transcription factors. Conversely,executioners of apoptosis such as Bax may be not functional, inclining thebalance towards inappropriate survival of the tumor cell. In breast cancer,many of these proteins are dysregulated. BID, Bcl-2 homology domain 3interacting domain death agonist; TNF, tumor necrosis factor; TRAIL,TNF-related apoptosis-inducing ligand.
Clinically explored strategies
Preliminary attempts to explore the therapeutic modulation of apoptosis against cancer by gene transfer have been started, driven by encouraging preclinical data in animal models. Clinical trials are currently ongoing to evaluate the value of pro-apoptotic p53 and adenoviral E1A, and a growing number of other candidate genes are being considered and tested preclinically (see [20,81,82] and Table 1).
Supplementation or restoration of p53
Direct induction of apoptosis has been attempted by replacing thetumor suppressor gene p53. Several factors made this gene anattractive candidate, including its frequent inactivation in human tumors, theobserved lack of toxicity of wild-type p53 itself, the control that itexerts in multiple other genes implicated in apoptosis and cell cycleregulation, and some experimental evidence for a bystander effect. Both invitro and in vivo animal studies [84,85,86,87] haveindeed shown induction of apoptosis and suppression of tumorigenicity in humanbreast cancer models after p53 gene delivery via recombinantadenoviral vectors. Of interest, this result has also been observed in tumorcell sublines selected for resistance to drugs that are commonly used in thetreatment of breast cancer [88] and in tumor cellsexpressing wild-type p53 [89]. Furthermore,adenovirus-mediated delivery of p53 augments the cytotoxic effect ofthe chemotherapeutic drug paclitaxel [90]. Thesepreclinical studies were followed by two clinical trials in breast cancer inwhich p53 is currently being administered intralesionally, orincubated ex vivo with bone marrow for purging of contaminating breastcancer cells [91]. Potential obstacles for thesuccessful clinical exploitation of p53, however, have arisen with theobservation that wild-type p53 can be inactivated in human breastcancer cells that express mutant p53 [92], andwith the low efficiency in vivo of current gene delivery vectors.
Adenoviral E1A
In addition to restoration of p53, the otherpro-apoptotic approach currently being clinically tested is based onliposome-mediated delivery of the adenoviral gene E1A [91,93]. Gene transfer of E1Ainhibits transcription of the human HER-2/neu promoter, and suppresses thetumorigenicity and metastatic potential of the HER-2/neu oncogene in cells thatoverexpress this oncogene [94]. An analogous effect ofE1A in anchorage-independent growth and tumorigenicity has been shownin a HER-2/neu-independent manner [95]. Finally,E1A sensitizes mammalian cells to immune-mediated apoptosis in ap53-independent manner [96]. Thus, delivery ofE1A could have an effect in vivo that exceeds the limitationsimposed by the heterogeneity in the HER-2/neu or p53 status within thetumor.
Requirements for pro-apoptotic gene therapy
A variety of additional interventions and targets have been proposed for the gene-based therapeutic induction of apoptosis (see Table 1, and the references therein). Here, comments are limited to theoretical aspects that need to be considered for designing successful therapeutic interventions for breast cancer based on the genetic modulation of apoptosis. Specifically, the gene transfer systems and knowledge of the genetic pathophysiology of the disease as the most critical aspects are considered.
High levels of gene transfer
Gene transfer vectors are needed that can trigger apopto-sis inmost malignant cells in any given tumor. Most frequently, current vectorsystems have been employed for delivery of therapeutic nucleic acids torelevant target cells in loco-regional contexts. In general, a fundamentalrecognition in most of these studies has been the disparity noted between thein vitro and in vivo gene transfer efficiencies of thesevectors, with a generally suboptimal tumor transduction [97]. For disseminated disease, employment of available vectorsis not presently feasible. This restriction is based on their limited capacityto accomplish efficient gene transfer to widely disseminated tumor targetsin vivo. Implicit in this limitation is the recognition of threerequirements for any candidate vector system for this purpose: the ability toaccomplish highly efficient and nontoxic gene delivery after direct invivo delivery via the intravascular route; the ability to accomplishtargeted, specific gene delivery to a selected cellular subset; and the abilityto escape the innate and adaptive immune responses.
With regard to the first requirement, only two presently availablevector systems, cationic liposomes and recombinant adenoviral vectors, havebeen reported to transduce various end organs after in vivo genedelivery [98,99,100,101]. However, low levels of geneexpression [102,103], orpromiscuous tropism and entrapment of the vector by the reticuloendothelialsystem, mostly in the liver [104], respectively, haveundermined the utility of the aforementioned vector systems for accomplishingtransduction of normal breast tissue and disseminated breast cancer [105]. This fundamental limit has hindered achieving thesecond requirement (ie the evaluation of tissue-specific or tumor-specificpromoters for transcriptional targeting of therapeutic genes in the context ofin vivo models of breast cancer) [105]. Withregard to the effects of the immune response, viral vectors display foreignantigens that can induce a strong cellular and humoral immune response inhumans [106,107,108]. This can ultimately determine immune-mediated clearanceof the vector and of the virally infected target cells [109], and precludes efficient readministration of thevector.
Thus, important limitations of current approaches used forimplementation of pro-apoptotic gene therapy for disseminated cancers,including breast cancer, have been noted. Although many potentially effectivestrategies exist to achieve the molecular treatment of breast cancer, genedelivery issues have limited definitive evaluation of these methods inclinically relevant models.
In this regard, understanding of the determinants of vectorefficacy at a cellular level has recently allowed vectors to be designed thatachieve cell-specific gene delivery in the loco-regional context. For instance,altering the tropism of adenoviral vectors by bifunctional, antibody-basedconjugates and by genetic engineering of the binding sites of the virusdramatically enhances the infectivity of otherwise refractory tumor cells[110]. This occurs by allowing the cellular entry ofthe virus through heterologous pathways, thus overcoming the paucity of itsreceptor, coxsackie and adenovirus receptor (CAR), which characterizes humanprimary tumors. Here the mandate for a high level of gene transfer may bestricter, given the lack of firm evidence for a useful bystander effectmediated by pro-apoptotic interventions, perhaps with the exception ofreplacement of p53 [111]. In this case, abetter definition of the basis of bystander effects observed after genedelivery of p53 should lead to the exploitation of similar schemas bydesigning ad hoc therapeutic payloads. Examples would be secretorymolecules such as the IGF binding protein 3 [112],which is induced by p53 and inhibits the powerful growth factor IGF-1;and translocating molecules, such as fusion proteins derived from the herpessimplex virus protein VP22 [113] or from the Tatprotein of human immunodeficiency virus type 1 [114,115,116,117]. Further advances are alsoneeded to understand the determinants of the biodistribution of viral vectorafter systemic intravascular administration. To this end, novel molecular andimaging technologies [118] have been developed thatallow the amount of vector in relevant tissues to be quantified [119] and gene expression to be followed noninvasively invivo [120*], both of which should accelerate thesefundamental studies.
High therapeutic index
Given the ubiquity of the numerous cellular proteins involved inapoptotic pathways, highly selective activation in cancer cells of lethalprocesses may also be a critical requirement of therapeutic maneuvers. Thelower threshold for undergoing apoptosis that characterizes tumor cells [1,2] could, however, offer anadvantageous therapeutic window that makes this requirement less stringent.Certain pro-apoptotic interventions might still require a more stringent levelof specificity. In such cases, vector targeting could provide the requiredrestriction in the expression of the pro-apoptotic molecules.
Targeted gene therapy for breast cancer can be accomplished atdifferent levels [121]. In one approach, the tumor cellcan be targeted at the level of transduction to achieve the selective deliveryof the therapeutic gene. This involves the derivation of a vector that bindsselectively to the target breast cancer cell. Alternatively, the therapeuticgene can be placed under the control of breast tumor-specific transcriptionalregulatory sequences that are activated in tumor cells, but not in normalcells, and therefore target expression selectively to the tumor cell (for areview see [28]). In addition, targeted gene therapy forcancer can exploit the unique physiology of solid tumors, such as hypoxia[122]. To date, targeted gene therapy has beenattempted by employing either transductional targeting or transcriptionaltargeting alone. Again, enhancement of the overall level of specificity bycombining the complementary approaches of transductional and transcriptionaltargeting may be required, each of which might be imperfect or'leaky' by itself [121]. Perhaps moreimportantly, vector targeting may confer, in addition to selectivity, asignificantly higher level of gene transfer, as mentioned above.
Multiple interventions and modalities
The signaling circuits that control apoptosis are complex andredundant, and tumors are formidably heterogeneous. Therefore, concomitantmodulation of several components of the apoptotic pathways may be needed toprovoke cell death in a robust and consistent manner. In this regard, thecomplex phenotype of the caspase knockout mice revealed that multiple andredundant mechanisms of caspase activation operate in parallel [123]. Recent high-throughput studies (for instance employingcomplementary DNA microarrays and serial analysis of gene expression) allowidentification of patterns of gene expression and their variation underappropriate stimuli, which will contribute to the dissection of the relevantpathways in breast cancer [124**,125]. Interventions downstream in the circuits might also bepreferable for avoiding regulatory counterbalances that may dissipate theeffect of the intervention. For instance, multiple genes act downstream ofp53 that alter the response to p53 restoration. In contrast,Bax has a more downstream role in effecting apoptosis, which perhaps explainsits more predictable cytotoxicity on tumor cells [126*].Finally, the magnitude of cell death after most pro-apoptotic interventionsin vitro is cell line-dependent, with some tumor cells typicallyshowing refractoriness to the induction of apoptosis. This has prompted theevaluation of combining a variety of pro-apoptotic interventions, and theirassociation with chemotherapy [4**], radiotherapy [6,127], and other biologicals [128]. These multimodality treatments have consistently showedthat higher levels of cell death are obtained when different pathways aremodulated and modalities are combined, clearly indicating that the targetedpathways do not totally overlap.
Conclusion
The contribution of a dysregulation of apoptosis to the malignant phenotype is increasingly being recognized. In that context, novel technologies for high throughput analysis of the expression profile of breast tissues, normal and malignant, under a variety of experimental and treatment conditions, are becoming widespread. The information thus collected will allow the apoptotic machinery and its dysfunction to be defined with extraordinary breadth and precision. At the same time, more efficient and selective gene transfer systems are being designed, which will allow the implementation of complex genetic interventions tailored to breast cancer and aimed at different components of the redundant regulatory pathways. The combination of such genetic modifications is most likely to be effective when combined with conventional treatments, thus robustly activating several pro-apoptotic pathways, and paving the way for the therapeutically beneficial application of pro-apoptotic gene therapy for cancer of the breast.
References
Evan G, Littlewood T: A matter of life and cell death. Science. 1998, 281: 1317-1322. 10.1126/science.281.5381.1317.
Fisher DE: Apoptosis in cancer therapy: crossing the threshold. Cell. 1994, 78: 539-542.
Sumantran VN, Ealovega MW, Nunez G, Clarke MF, Wicha MS: Overexpression of Bcl-XS sensitizes MCF-7 cells to chemotherapy-induced apoptosis. Cancer Res. 1995, 55: 2507-2510.
Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S: Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999, 59: 734-741.
Muschel RJ, Soto DE, McKenna WG, Bernhard EJ: Radiosensitization and apoptosis. Oncogene. 1998, 17: 3359-3363. 10.1038/sj.onc.1202580.
Sheard MA, Krammer PH, Zaloudik J: Fractionated gamma-irradiation renders tumour cells more responsive to apoptotic signals through CD95. Br J Cancer. 1999, 80: 1689-1696. 10.1038/sj.bjc.6690585.
Kerr JF, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972, 26: 239-257.
Kerr JF, Winterford CM, Harmon BV: Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994, 73: 2013-2026.
Williams GT: Programmed cell death: apoptosis and oncogenesis. Cell. 1991, 65: 1097-1098.
Strasser A: Dr. Josef Steiner Cancer Research Prize Lecture: the role of physiological cell death in neoplastic transformation and in anti-cancer therapy. Int J Cancer. 1999, 81: 505-511. 10.1002/(SICI)1097-0215(19990517)81:4<505::AID-IJC1>3.0.CO;2-J.
Allan DJ, Howell A, Roberts SA, et al: Reduction in apoptosis relative to mitosis in histologically normal epithelium accompanies fibrocystic change and carcinoma of the premenopausal human `breast. J Pathol. 1992, 167: 25-32.
Shibata MA, Liu ML, Knudson MC, et al: Haploid loss of bax leads to accelerated mammary tumor development in C3(1)/SV40-TAg transgenic mice: reduction in protective apoptotic response at the preneoplastic stage. EMBO J. 1999, 18: 2692-2701. 10.1093/emboj/18.10.2692.
Jaattela M, Benedict M, Tewari M, Shayman JA, Dixit VM: Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene. 1995, 10: 2297-2305.
Frisch SM: Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr Biol . 1999, 9: 1047-1049. 10.1016/S0960-9822(99)80455-2.
Hannun YA: Apoptosis and the dilemma of cancer chemotherapy. Blood. 1997, 89: 1845-1853.
Haimovitz-Friedman A, Kan CC, Ehleiter D, et al: Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994, 180: 525-535.
Zhang GJ, Kimijima I, Onda M, et al: Tamoxifen-induced apoptosis in breast cancer cells relates to down-regulation of bcl-2, but not bax and bcl-X(L), without alteration of p53 protein levels. Clin Cancer Res. 1999, 5: 2971-2977.
Shao ZM, Li J, Wu J, et al: Neo-adjuvant chemotherapy for operable breast cancer induces apoptosis. Breast Cancer Res Treat. 1999, 53: 263-269. 10.1023/A:1006194921139.
Reed JC: Bcl-2: prevention of apoptosis as a mechanism of drug resistance. Hematol Oncol Clin North Am. 1995, 9: 451-473.
Schmitt CA, Lowe SW: Apoptosis and therapy. J Pathol . 1999, 187: 127-137. 10.1002/(SICI)1096-9896(199901)187:1<127::AID-PATH251>3.0.CO;2-T.
Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B: The biochemistry of programmed cell death. FASEB J. 1995, 9: 1277-1287.
Walker RA, Jones JL, Chappell S, Walsh T, Shaw JA: Molecular pathology of breast cancer and its application to clinical management. Cancer Metastasis Rev. 1997, 16: 5-27. 10.1023/A:1005740222307.
Wu J: Apoptosis and angiogenesis: two promising tumor markers in breast cancer. Anticancer Res. 1996, 16: 2233-2239.
Brenner AJ, Aldaz CM: The genetics of sporadic breast cancer. Prog Clin Biol Res. 1997, 396: 63-82.
Dickson RB, Lippman ME: Growth factors in breast cancer. Endocr Rev. 1995, 16: 559-589. 10.1210/er.16.5.559.
Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB: Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996, 2: 1215-1219.
Gooch JL, Van Den Berg CL, Yee D: Insulin-like growth factor (IGF)-1 rescues breast cancer cells from chemotherapy-induced cell death: proliferative and anti-apoptotic effects. Breast Cancer Res Treat . 1999, 56: 1-10. 10.1023/A:1006208721167.
Ruppert JM, Wright M, Rosenfeld M, et al: Gene therapy strategies for carcinoma of the breast. Breast Cancer Res Treat . 1997, 44: 93-114. 10.1023/A:1005761723853.
Boxhorn HK, Eck SL: Gene therapy for breast cancer. Hematol Oncol Clin North Am. 1998, 12: 665-675.
Elledge RM, Allred DC: The p53 tumor suppressor gene in breast cancer. Breast Cancer Res Treat. 1994, 32: 39-47.
Berns EM, De Witte HH, Klijn JG, et al: Prognostic value of TP53 protein accumulation in human primary breast cancer: an analysis by luminometric immunoassay on 1491 tumor cytosols. Anticancer Res. 1997, 17: 3003-3006.
White E: Tumour biology. p53, guardian of Rb. Nature . 1994, 371: 21-22. 10.1038/371021a0.
Livingstone LR, White A, Sprouse J, et al: Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992, 70: 923-935.
Sheikh MS, Rochefort H, Garcia M: Overexpression of p21WAF1/CIP1 induces growth arrest, giant cell formation and apoptosis in human breast carcinoma cell lines. Oncogene. 1995, 11: 1899-1905.
Sierra A, Lloveras B, Castellsague X, et al: Bcl-2 expression is associated with lymph node metastasis in human ductal breast carcinoma. Int J Cancer. 1995, 60: 54-60.
Eissa S, Labib R, Khalifa A, et al: Regulators of apoptosis in human breast cancer. Clin Biochem. 1999, 32: 321-326. 10.1016/S0009-9120(99)00025-9.
Mustonen M, Raunio H, Paakko P, Soini Y: The extent of apoptosis is inversely associated with bcl-2 expression in premalignant and malignant breast lesions. Histopathology. 1997, 31: 347-354. 10.1046/j.1365-2559.1997.2710877.x.
Holmqvist P, Lundstrom M, Stal O: Apoptosis and Bcl-2 expression in relation to age, tumor characteristics and prognosis in breast cancer. South-East Sweden Breast Cancer Group. Int J Biol Markers. 1999, 14: 84-91.
Sierra A, Castellsague X, Coll T, et al: Expression of death-related genes and their relationship to loss of apoptosis in T1 ductal breast carcinomas. Int J Cancer. 1998, 79: 103-110. 10.1002/(SICI)1097-0215(19980417)79:2<103::AID-IJC1>3.0.CO;2-X.
Sierra A, Castellsague X, Coll T, et al: Expression of death-related genes and their relationship to loss of apoptosis in T1 ductal breast carcinomas. Int J Cancer. 1998, 79: 103-110. 10.1002/(SICI)1097-0215(19980417)79:2<103::AID-IJC1>3.0.CO;2-X.
Leek RD, Kaklamanis L, Pezzella F, Gatter KC, Harris AL: bcl-2 in normal human breast and carcinoma, association with oestrogen receptor-positive, epidermal growth factor receptor-negative tumours and in situ cancer. Br J Cancer. 1994, 69: 135-139.
Joensuu H, Pylkkanen L, Toikkanen S: Bcl-2 protein expression and long-term survival in breast cancer. Am J Pathol. 1994, 145: 1191-1198.
Silvestrini R, Veneroni S, Daidone MG, et al: The Bcl-2 protein: a prognostic indicator strongly related to p53 protein in lymph node-negative breast cancer patients. J Natl Cancer Inst. 1994, 86: 499-504.
Zhang GJ, Kimijima I, Abe R, et al: Apoptotic index correlates to bcl-2 and p53 protein expression, histological grade and prognosis in invasive breast cancers. Anticancer Res. 1998, 18: 1989-1998.
van Slooten HJ, van de Vijver MJ, van de Velde CJ, van Dierendonck JH: Loss of Bcl-2 in invasive breast cancer is associated with high rates of cell death, but also with increased proliferative activity. Br J Cancer. 1998, 77: 789-796.
Rochaix P, Krajewski S, Reed JC, et al: In vivo patterns of Bcl-2 family protein expression in breast carcinomas in relation to apoptosis. J Pathol. 1999, 187: 410-415. 10.1002/(SICI)1096-9896(199903)187:4<410::AID-PATH266>3.0.CO;2-F.
Vakkala M, Lahteenmaki K, Raunio H, Paakko P, Soini Y: Apoptosis during breast carcinoma progression. Clin Cancer Res. 1999, 5: 319-324.
Hurlimann J, Larrinaga B, Vala DL: bcl-2 protein in invasive ductal breast carcinomas. Virchows Arch. 1995, 426: 163-168.
Gee JM, Robertson JF, Ellis IO, et al: Immunocytochemical localization of BCL-2 protein in human breast cancers and its relationship to a series of prognostic markers and response to endocrine therapy. Int J Cancer. 1994, 59: 619-628.
Gasparini G, Barbareschi M, Doglioni C, et al: Expression of bcl-2 protein predicts efficacy of adjuvant treatments in operable node-positive breast cancer. Clin Cancer Res. 1995, 1: 189-198.
Olopade OI, Adeyanju MO, Safa AR, et al: Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997, 3: 230-237.
Krajewski S, Blomqvist C, Franssila K, et al: Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995, 55: 4471-4478.
Veronese S, Mauri FA, Caffo O, et al: Bax immunohistochemical expression in breast carcinoma: a study with long term follow-up. Int J Cancer. 1998, 79: 13-18. 10.1002/(SICI)1097-0215(19980220)79:1<13::AID-IJC3>3.0.CO;2-Z.
Zhan Q, Fan S, Bae I, et al: Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene . 1994, 9: 3743-3751.
Bargou RC, Wagener C, Bommert K, et al: Overexpression of the death-promoting gene bax-alpha which is downregulated in breast cancer restores sensitivity to different apoptotic stimuli and reduces tumor growth in SCID mice. J Clin Invest. 1996, 97: 2651-2659.
Perez GI, Knudson CM, Leykin L, Korsmeyer SJ, Tilly JL: Apoptosis-associated signaling pathways are required for chemotherapy-mediated female germ cell destruction. Nature Med . 1997, 3: 1228-1232.
Oltvai ZN, Milliman CL, Korsmeyer SJ: Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993, 74: 609-619.
**ang J, Chao DT, Korsmeyer SJ: BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996, 93: 14559-14563. 10.1073/pnas.93.25.14559.
Marzo I, Brenner C, Zamzami N, et al: Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998, 281: 2027-2031. 10.1126/science.281.5385.2027.
Thornberry NA, Lazebnik Y: Caspases: enemies within. Science. 1998, 281: 1312-1316. 10.1126/science.281.5381.1312.
Wallach D, Varfolomeev EE, Malinin NL, et al: Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999, 17: 331-367. 10.1146/annurev.immunol.17.1.331.
Li H, Zhu H, Xu CJ, Yuan J: Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998, 94: 491-501.
Yin XM, Wang K, Gross A, et al: Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999, 400: 886-891. 10.1038/23730.
Gross A, Yin XM, Wang K, et al: Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999, 274: 1156-1163. 10.1074/jbc.274.2.1156.
Muschen M, Moers C, Warskulat U, et al: CD95 ligand expression in dedifferentiated breast cancer. J Pathol. 1999, 189: 378-386. 10.1002/(SICI)1096-9896(199911)189:3<378::AID-PATH439>3.0.CO;2-D.
Jaattela M: Esca** cell death: survival proteins in cancer. Exp Cell Res. 1999, 248: 30-43. 10.1006/excr.1999.4455.
LaCasse EC, Baird S, Korneluk RG, MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998, 17: 3247-3259. 10.1038/sj.onc.1202569.
Deveraux QL, Reed JC: IAP family proteins: suppressors of apoptosis. Genes Dev. 1999, 13: 239-252.
Ambrosini G, Adida C, Altieri DC: A novel anti-apoptosis gene, surviving, expressed in cancer and lymphoma. Nature Med. 1997, 3: 917-921.
Tamm I, Wang Y, Sausville E, et al: IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58: 5315-5320.
Li F, Ambrosini G, Chu EY, et al: Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998, 396: 580-584. 10.1038/25141.
Ciocca DR, Clark GM, Tandon AK, et al: Heat shock protein hsp70 in patients with axillary lymph node-negative breast cancer: prognostic implications. J Natl Cancer Inst. 1993, 85: 570-574.
Lazaris AC, Chatzigianni EB, Panoussopoulos D, et al: Proliferating cell nuclear antigen and heat shock protein 70 immunolocalization in invasive ductal breast cancer not otherwise specified. Breast Cancer Res Treat. 1997, 43: 43-51. 10.1023/A:1005706110275.
Vargas-Roig LM, Fanelli MA, Lopez LA, et al: Heat shock proteins and cell proliferation in human breast cancer biopsy samples. Cancer Detect Prev. 1997, 21: 441-451.
Vargas-Roig LM, Gago FE, Tello O, Aznar JC, Ciocca DR: Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int J Cancer. 1998, 79: 468-475. 10.1002/(SICI)1097-0215(19981023)79:5<468::AID-IJC4>3.0.CO;2-Z.
Liu FF, Miller N, Levin W, et al: The potential role of HSP70 as an indicator of response to radiation and hyperthermia treatments for recurrent breast cancer. Int J Hyperthermia. 1996, 12: 197-208.
Ciocca DR, Green S, Elledge RM, et al: Heat shock proteins hsp27 and hsp70: lack of correlation with response to tamoxifen and clinical course of disease in estrogen receptor-positive metastatic breast cancer (a Southwest Oncology Group Study). Clin Cancer Res. 1998, 4: 1263-1266.
Green DR, Reed JC: Mitochondria and apoptosis. Science . 1998, 281: 1309-1312. 10.1126/science.281.5381.1309.
Ashkenazi A, Dixit VM: Death receptors: signaling and modulation. Science. 1998, 281: 1305-1308. 10.1126/science.281.5381.1305.
Thornberry NA, Lazebnik Y: Caspases: enemies within. Science. 1998, 281: 1312-1316. 10.1126/science.281.5381.1312.
Favrot M, Coll JL, Louis N, Negoescu A: Cell death and cancer: replacement of apoptotic genes and inactivation of death suppressor genes in therapy. Gene Ther. 1998, 5: 728-739. 10.1038/sj.gt.3300661.
Reed JC: Apoptosis as a goal of cancer gene therapy. In: The Development of Human Gene Therapy. Edited by Friedmann T. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. 1999, : 545-572.
Neubauer A, Thiede C, Huhn D, Wittig B: P53 and induction of apoptosis as a target for anticancer therapy [Review]. Leukemia. 1996, 10 (suppl 3): S2-S4.
Nielsen LL, Maneval DC: P53 tumor suppressor gene therapy for cancer. Cancer Gene Ther. 1998, 5: 52-63.
Seth P, Brinkmann U, Schwartz GN, et al: Adenovirus-mediated gene transfer to human breast tumor cells: an approach for cancer gene therapy and bone marrow purging. Cancer Res. 1996, 56: 1346-1351.
Nielsen LL, Dell J, Maxwell E, et al: Efficacy of p53 adenovirus-mediated gene therapy against human breast cancer xenografts. Cancer Gene Ther. 1997, 4: 129-138.
Nielsen LL, Gurnani M, Syed J, et al: Recombinant E1-deleted adenovirus-mediated gene therapy for cancer: efficacy studies with p53 tumor suppressor gene and liver histology in tumor xenograft models. Hum Gene Ther. 1998, 9: 681-694.
Seth P, Katayose D, Li Z, et al: A recombinant adenovirus expressing wild type p53 induces apoptosis in drug-resistant human breast cancer cells: a gene therapy approach for drug-resistant cancers. Cancer Gene Ther. 1997, 4: 383-390.
Li P, Bui T, Gray D, Klamut HJ: Therapeutic potential of recombinant p53 overexpression in breast cancer cells expressing endogenous wild-type p53. Breast Cancer Res Treat. 1998, 48: 273-286. 10.1023/A:1005961705860.
Nielsen LL, Lipari P, Dell J, Gurnani M, Hajian G: Adenovirus-mediated p53 gene therapy and paclitaxel have synergistic efficacy in models of human head and neck, ovarian, prostate, and breast cancer. Clin Cancer Res. 1998, 4: 835-846.
Office of Recombinant DNA Activities. 1998, [http://www.nih.gov/od/orda/docs.pdf]
Vinyals A, Peinado MA, Gonzalez-Garrigues M, et al: Failure of wild-type p53 gene therapy in human cancer cells expressing a mutant p53 protein. Gene Ther. 1999, 6: 22-33. 10.1038/sj.gt.3300786.
Hortobagyi GN, Hung MC, Lopez-Berestein G: A Phase I multicenter study of E1A gene therapy for patients with metastatic breast cancer and epithelial ovarian cancer that overexpresses HER-2/neu or epithelial ovarian cancer. Hum Gene Ther. 1998, 9: 1775-1798.
Chang JY, **a W, Shao R, et al: The tumor suppression activity of E1A in HER-2/neu-overexpressing breast cancer. Oncogene . 1997, 14: 561-568. 10.1038/sj.onc.1200861.
Frisch SM, Dolter KE: Adenovirus E1a-mediated tumor suppression by a c-erbB-2/neu-independent mechanism. Cancer Res. 1995, 55: 5551-5555.
Cook JL, Routes BA, Leu CY, Walker TA, Colvin KL: E1A oncogene-induced cellular sensitization to immune-mediated apoptosis is independent of p53 and resistant to blockade by E1B 19 kDa protein. Exp Cell Res. 1999, 252: 199-210. 10.1006/excr.1999.4617.
Takamiya Y, Short MP, Ezzeddine ZD, et al: Gene therapy of malignant brain tumors: a rat glioma line bearing the herpes simplex virus type 1-thymidine kinase gene and wild type retrovirus kills other tumor cells. J Neurosci Res. 1992, 33: 493-503.
Debs RJ, Heath TD, Papahadjopoulos D: Targeting of anti-Thy 1.1 monoclonal antibody conjugated liposomes in Thy 1.1 mice after intravenous administration. Biochim Biophys Acta. 1987, 901: 183-190. 10.1016/0005-2736(87)90114-3.
Stratford-Perricaudet LD, Briand P, Perricaudet M: Feasibility of adenovirus-mediated gene transfer in vivo. Bone Marrow Transplant. 1992, 9 (suppl 1): 151-152.
Brody SL, Crystal RG: Adenovirus-mediated in vivo gene transfer. Ann N Y Acad Sci. 1994, 716: 90-101.
Heise C, Sampson-Johannes A, Williams A, et al: ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nature Med. 1997, 3: 639-645.
Lesoon-Wood LA, Kim WH, Kleinman HK, Weintraub BD, Mixson AJ: Systemic gene therapy with p53 reduces growth and metastases of a malignant human breast cancer in nude mice. Hum Gene Ther. 1995, 6: 395-405.
Chen QR, Mixson JA: Systemic gene therapy with p53 inhibits breast cancer: recent advances and therapeutic implications. Front Biosci. 1998, 3: D997-D1004.
Vrancken PM, Perkins AL, Kay MA: Method for multiple portal vein infusions in mice: quantitation of adenovirus-mediated hepatic gene transfer. Biotechniques. 1996, 20: 278-285.
Anderson LM, Swaminathan S, Zackon I, et al: Adenovirus-mediated tissue-targeted expression of the HSVtk gene for the treatment of breast cancer. Gene Ther. 1999, 6: 854-864. 10.1038/sj.gt.3300909.
Molnar-Kimber KL, Sterman DH, Chang M, et al: Impact of preexisting and induced humoral and cellular immune responses in an adenovirus-based gene therapy phase I clinical trial for localized mesothelioma. Hum Gene Ther. 1998, 9: 2121-2133.
Gahery-Segard H, Farace F, Godfrin D, et al: Immune response to recombinant capsid proteins of adenovirus in humans: antifiber and anti-penton base antibodies have a synergistic effect on neutralizing activity. J Virol. 1998, 72: 2388-2397.
Elkon KB, Liu CC, Gall JG, et al: Tumor necrosis factor alpha plays a central role in immune-mediated clearance of adenoviral vectors. Proc Natl Acad Sci USA. 1997, 94: 9814-9819. 10.1073/pnas.94.18.9814.
Kafri T, Morgan D, Krahl T, et al: Cellular immune response to adenoviral vector infected cells does not require de novo viral gene expression: implications for gene therapy. Proc Natl Acad Sci USA. 1998, 95: 11377-11382. 10.1073/pnas.95.19.11377.
Reynolds PN, Curiel DT: Strategies to adapt adenoviral vectors for gene therapy applications: targeting and integration. In: The Development of Human Gene Therapy. Edited by Friedmann T. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. 1999, : 111-130.
Bouvet M, Ellis LM, Nishizaki M, et al: Adenovirus-mediated wild-type p53 gene transfer down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human colon cancer. Cancer Res. 1998, 58: 2288-2292.
Buckbinder L, Talbott R, Velasco-Miguel S, et al: Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995, 377: 646-649. 10.1038/377646a0.
Dilber MS, Phelan A, Aints A, et al: Intercellular delivery of thymidine kinase prodrug activating enzyme by the herpes simplex virus protein, VP22. Gene Ther. 1999, 6: 12-21. 10.1038/sj.gt.3300838.
Fawell S, Seery J, Daikh Y, et al: Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994, 91: 664-668.
Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF: In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999, 285: 1569-1572. 10.1126/science.285.5433.1569.
Phelan A, Elliott G, O'Hare P: Intercellular delivery of functional p53 by the herpesvirus protein VP22. Nature Biotechnol . 1998, 16: 440-443.
Elliott G, O'Hare P: Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997, 88: 223-233.
Gambhir SS, Barrio JR, Herschman HR, Phelps ME: Assays for non-invasive imaging of reporter gene expression. Nucl Med Biol. 1999, 26: 481-490. 10.1016/S0969-8051(99)00021-9.
Becker K, Pan D, Whitley CB: Real-time quantitative polymerase chain reaction to assess gene transfer. Hum Gene Ther. 1999, 10: 2559-2566. 10.1089/10430349950016898.
Rogers BE, McLean SF, Kirkman RL, et al: In vivo localization of [(111)In]-DTPA-D-Phe1-octreotide to human ovarian tumor xenografts induced to express the somatostatin receptor subtype 2 using an adenoviral vector. Clin Cancer Res. 1999, 5: 383-393.
Douglas JT, Curiel DT: Targeted gene therapy. Tumor Targeting. 1995, 2: 67-84.
Binley K, Iqball S, Kingsman A, Kingsman S, Naylor S: An adenoviral vector regulated by hypoxia for the treatment of ischaemic disease and cancer. Gene Ther. 1999, 6: 1721-1727. 10.1038/sj.gt.3301001.
Li H, Yuan J: Deciphering the pathways of life and death. Curr Opin Cell Biol. 1999, 11: 261-266. 10.1016/S0955-0674(99)80035-0.
Perou CM, Jeffrey SS, van de RM, et al: Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci USA. 1999, 96: 9212-9217. 10.1073/pnas.96.16.9212.
Nacht M, Ferguson AT, Zhang W, et al: Combining serial analysis of gene expression and array technologies to identify genes differentially expressed in breast cancer. Cancer Res. 1999, 59: 5464-5470.
**ang J, Piche A, Rancourt C, et al: Adenoviral vector-mediated expression of Bax selectively induces apoptosis in ovarian cancer cells. Tumor Targeting. 1999, 4: 84-91.
Zhivotovsky B, Joseph B, Orrenius S: Tumor radiosensitivity and apoptosis. Exp Cell Res. 1999, 248: 10-17. 10.1006/excr.1999.4452.
Putzer BM, Bramson JL, Addison CL, et al: Combination therapy with interleukin-2 and wild-type p53 expressed by adenoviral vectors potentiates tumor regression in a murine model of breast cancer. Hum Gene Ther. 1998, 9: 707-718.
Paillard F: Induction of apoptosis with I-kappaB, the inhibitor of NF-kappaB. Hum Gene Ther. 1999, 10: 1-3. 10.1089/10430349950019138.
Rakkar AN, Katayose Y, Kim M, et al: A novel adenoviral vector expressing human Fas/CD95/APO-1 enhances p53-mediated apoptosis. Cell Death Differ. 1999, 6: 326-333. 10.1038/sj.cdd.4400498.
Wright M, Grim J, Deshane J, et al: An intracellular anti-erbB-2 single-chain antibody is specifically cytotoxic to human breast carcinoma cells overexpressing erbB-2. Gene Ther. 1997, 4: 317-322. 10.1038/sj.gt.3300372.
Piche A, Grim J, Rancourt C, et al: Modulation of Bcl-2 protein levels by an intracellular anti-Bcl-2 single-chain antibody increases drug-induced cytotoxicity in the breast cancer cell line MCF-7. Cancer Res. 1998, 58: 2134-2140.
Ambrosini G, Adida C, Sirugo G, Altieri DC: Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting. J Biol Chem. 1998, 273: 11177-11182. 10.1074/jbc.273.18.11177.
Wei Y, Zhao X, Kariya Y, Teshigawara K, Uchida A: Inhibition of proliferation and induction of apoptosis by abrogation of heat-shock protein (HSP) expression in tumor cells. Cancer Immunol Immunother. 1995, 40: 73-78. 10.1007/s002620050146.
Su ZZ, Madireddi MT, Lin JJ, et al: The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proc Natl Acad Sci USA. 1998, 95: 14400-14405. 10.1073/pnas.95.24.14400.
Tai YT, Strobel T, Kufe D, Cannistra SA: In vivo cytotoxicity of ovarian cancer cells through tumor-selective expression of the BAX gene. Cancer Res. 1999, 59: 2121-2126.
Clarke MF, Apel IJ, Benedict MA, et al: A recombinant bcl-xs adenovirus selectively induces apoptosis in cancer cells but not in normal bone marrow cells. Proc Natl Acad Sci USA. 1995, 92: 11024-11028.
Marcelli M, Cunningham GR, Walkup M, et al: Signaling pathway activated during apoptosis of the prostate cancer cell line LNCaP: overexpression of caspase-7 as a new gene therapy strategy for prostate cancer. Cancer Res. 1999, 59: 382-390.
Ealovega MW, McGinnis PK, Sumantran VN, Clarke MF, Wicha MS: bcl-xs gene therapy induces apoptosis of human mammary tumors in nude mice. Cancer Res. 1996, 56: 1965-1969.
Danen-Van Oorschot AA, Fischer DF, Grimbergen JM, et al: Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci USA. 1997, 94: 5843-5847. 10.1073/pnas.94.11.5843.
Pietersen AM, van der Eb MM, Rademaker HJ, et al: Specific tumor-cell killing with adenovirus vectors containing the apoptin gene. Gene Ther. 1999, 6: 882-892. 10.1038/sj.gt.3300876.
Shao N, Chai YL, Shyam E, Reddy P, Rao VN: Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene. 1996, 13: 1-7.
Le XF, Vallian S, Mu ZM, Hung MC, Chang KS: Recombinant PML adenovirus suppresses growth and tumorigenicity of human breast cancer cells by inducing G1 cell cycle arrest and apoptosis. Oncogene. 1998, 16: 1839-1849. 10.1038/sj.onc.1201705.
Putzer BM, Bramson JL, Addison CL, et al: Combination therapy with interleukin-2 and wild-type p53 expressed by adenoviral vectors potentiates tumor regression in a murine model of breast cancer. Hum Gene Ther. 1998, 9: 707-718.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gómez-Navarro, J., Arafat, W. & **ang, J. Gene therapy for carcinoma of the breast: Pro-apoptotic gene therapy. Breast Cancer Res 2, 32 (1999). https://doi.org/10.1186/bcr27
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr27