
Abstract
Introduction
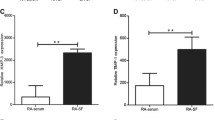
We previously demonstrated that a single-chain fragment variable (scFv) specific to collagen type II (CII) posttranslationally modified by reactive oxygen species (ROS) can be used to target anti-inflammatory therapeutics specifically to inflamed arthritic joints. The objective of the present study was to demonstrate the superior efficacy of anti-inflammatory cytokines when targeted to inflamed arthritic joints by the anti-ROS modified CII (anti-ROS-CII) scFv in a mouse model of arthritis.
Methods
Viral interleukin-10 (vIL-10) was fused to anti-ROS-CII scFv (1-11E) with a matrix-metalloproteinase (MMP) cleavable linker to create 1-11E/vIL-10 fusion. Binding of 1-11E/vIL-10 to ROS-CII was determined by enzyme-linked immunosorbent assay (ELISA), Western blotting, and immune-staining of arthritic cartilage, whereas vIL-10 bioactivity was evaluated in vitro by using an MC-9 cell-proliferation assay. Specific in vivo localization and therapeutic efficacy of 1-11E/vIL-10 was tested in the mouse model of antigen-induced arthritis.
Results
1-11E/vIL-10 bound specifically to ROS-CII and to damaged arthritic cartilage. Interestingly, the in vitro vIL-10 activity in the fusion protein was observed only after cleavage with MMP-1. When systemically administered to arthritic mice, 1-11E/vIL-10 localized specifically to the arthritic knee, with peak accumulation observed after 3 days. Moreover, 1-11E/vIL-10 reduced inflammation significantly quicker than vIL-10 fused to the control anti-hen egg lysozyme scFv (C7/vIL10).
Conclusions
Targeted delivery of anti-inflammatory cytokines potentiates their anti-arthritic action in a mouse model of arthritis. Our results further support the hypothesis that targeting biotherapeutics to arthritic joints may be extended to include anti-inflammatory cytokines that lack efficacy when administered systemically.
Similar content being viewed by others


Introduction
The hallmark of rheumatoid arthritis (RA) is chronic synovial inflammation that results in progressive joint damage. The pathogenesis of the disease is characterized by autoimmunity, chronic inflammatory synovitis, and destruction of the cartilage and the adjacent joint tissues [1]. These pathogenic processes are due to an imbalance in the cytokine network, where pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-1β, and IL-6 are overexpressed in the RA joint [2]. Homeostatic regulatory mechanisms in turn result in increased production of anti-inflammatory cytokines, such as IL-10 and transforming growth factor (TGF)-β, but this is not sufficient to counter the pro-inflammatory cytokines produced [3].
With this in mind, two alternative therapeutic approaches have been investigated. One is to neutralize the pro-inflammatory cytokines, and the other is to increase the concentration of the anti-inflammatory cytokines. Systemic treatment with TNF-α-blocking reagents is now a standard treatment of patients with RA failing to respond to conventional disease-modifying anti-rheumatic drugs (DMARDs) [4]. However, increasing evidence suggests that inhibition of TNF-α is associated with increased risk of infections due to general immune-suppression [5, 6]. Moreover, despite the established clinical efficacy of anti-TNF-α, a subset of patients (30% to 40%) does not respond or has a suboptimal clinical response to anti-TNF-α treatment [7].
Our hypothesis is that targeting of biologic drugs to the inflamed joint will result in high local concentrations and low systemic concentrations, increasing efficacy while minimizing side effects. Additionally, a lower total dose may be effective, thereby reducing the cost of treatment. Targeting could be achieved by the identification of an inflamed joint tissue specific marker. We hypothesized that the influx of infiltrating leukocytes consumes increased amounts of oxygen and thereby generates high levels of ROS [8]. In turn, the influx of ROS results in chemical posttranslational modification of major specific cartilage components such as CII, resulting in formation of ROS-CII. ROS-CII would therefore be present in inflamed joints, but not in healthy joints, and thus represents an inflamed joint-specific target. Targeting may therefore be achieved by an ROS-CII-specific antibody.
By using a phage display human antibody library, we have developed a panel of human single-chain fragment variable (scFv) that binds specifically to ROS-CII [9]. One of these clones, 1-11E, localizes specifically in arthritic joints of mice. Hence, 1-11E fused to the murine tumor necrosis factor receptor 2-Fc-fusion protein (mTNFR2-Fc), which would scavenge pro-inflammatory TNF-α, had an enhanced therapeutic effect on inflamed knee swelling compared with mTNFR2-Fc fused to the nonrelevant anti-hen egg lysozyme (HEL) scFv, (C7/mTNFR2-Fc), or mTNFR2-Fc alone.
The current study is built on the previous study with 1-11E/mTNFR2-Fc to extend the range of targeted therapeutics to include an anti-inflammatory cytokine, IL-10. IL-10 is a major anti-inflammatory cytokine that inhibits the production of Th1 cytokines, such as interferon-γ, Th17 cytokines, and IL-17 [Full size image
We performed transient transfections by using adherent HEK-293 cells using FuGENE 6 and according to manufacturer’s instructions (Promega UK, Southampton, UK). Culture medium was harvested after 3 days, and fusion protein was purified by using a nickel chelate column (QIAGEN, Crawley, UK) and following the manufacturer’s instructions.
Evaluation of the molecular integrity of the fusion protein
Fusion protein was separated on SDS-PAGE under denaturing conditions after purification with Ni-NTA Agarose (Qiagen, Crawley, UK) or after overnight digestion of 1-11E/vIL-10 fusion by the catalytic domains of MMP-1, MMP-3, and MMP-12 [19]. Western blotting with anti-penta-His HRP was performed as described [9]. Gel filtration chromatography was performed by using a HiPrep 16/60 sephacryl S-200 (GE Healthcare, Little Chalfont, UK) column connected to an AKTA FPLC (GE Healthcare) in sterile 20 mM Tris–HCl, 150 mM NaCl, pH 8.
In vitro bioactivity
Binding of purified fusion proteins to native CII and ROS-CII was determined by ELISA and Western blotting as described [9], with the bound fusion protein detected by using mouse anti-vIL-10 (R&D Systems, Abingdon, UK) followed by anti-mouse HRP (Sigma, Dorset, UK). Bioactivity of vIL-10 in the fusion protein was tested with a cell-proliferation assay by using mouse MC-9 mast cells as described [18]. In brief, on day 1, 5 × 104 cells were treated with fusion proteins (with or without MMP-1 digestion) at concentrations of 1,000 ng/ml, 100 ng/ml, or 10 ng/ml, as well as control recombinant vIL-10 (R&D Systems, Abingdon UK). On day 4, cell viability was measured with Cell Titer Glo (Promega, Southampton, UK), and plates were read in a luminometer (Dynex, Worthing, UK) as per manufacturer’s instructions.
Cartilage immunohistochemistry
Immunohistochemistry, by using damaged arthritic cartilage from several mouse models of arthritis, was performed. Samples were from C57BL/6 mice with antigen-induced arthritis (AIA) [20], DBA mice with collagen-induced arthritis (CIA) [19], and C57BL/6 with destabilization of the medial meniscus (DMM) [21]. All animal procedures were carried out under Home Office project licenses issued under the Animals (Scientific Procedures) Act 1986 as amended, and the institutional Animal Welfare and Ethical Review Bodies of Queen Mary University of London and University of Oxford, approved by the licenses from the Home Office. Immunohistochemistry was also performed by using human OA cartilage obtained from a patient undergoing prosthetic knee replacement (provided by Professor C. Montecucco, Fondazione IRCCS Policlinico S. Matteo, Pavia, Italy). Human OA were collected after consent, in accordance with institutional ethics policies and regulations (approved by Fondazione IRCCS Policlinico San Matteo, Pavia, Italy).
Safranin O staining was performed according to standard protocols [22]. Cartilage immunostaining was performed on 5-μm-thick sections, which were deparaffinized, hydrated, antigen retrieved, and blocked, as described [9]. Sections were incubated overnight at 4°C with 1-11E/vIL-10 or control C7/vIL-10 fusion (10 μg/ml) in DAKO diluent solution. Binding of the fusion protein was probed by using mouse anti-vIL-10 (R&D Systems, Abingdon, UK) followed by 1:1,000 anti-mouse HRP (Sigma, Dorset, UK). DAB substrate was used as peroxidase substrate (DAKO, Ely, Cambridgeshire, UK). Sections were counterstained with hematoxylin and mounted with DPX mount (BDH, London, UK). Fluorescent immunohistochemistry was also performed by using Cy5.5-labelled 1-11E/vIL-10 and C7/vIL-10. Cy5.5 labeling was done according to the manufacturer’s instructions (GE Healthcare, Buckinghamshire, UK). The pericellular matrix was stained by using the rat anti-heparan sulfate proteoglycan antibody (Millipore, Watford, UK) followed by the Alexa-Fluor-488 labeled anti-rat IgG (Life Technologies, Paisley, UK). Slides were viewed under the LSM 510 Meta (Zeiss, Cambridge, UK) using the 488-nm excitation laser to visualize the Alexa-488 label (green) and the 633-nm excitation laser to visualize the Cy5.5 label (red).
Mouse models of arthritis
Female C57BL/6 mice of 10 weeks of age were used for AIA, as described previously [20]. In brief, after initial immunization with 100 μg mBSA in complete Freud adjuvant, inflammation was induced in the knees by intraarticular injection of 50 μg mBSA in PBS.
CIA was induced as described [19]. In brief, 10-week-old male DBA/1 mice were immunized by intradermal injection of an emulsion of 200 μg of bovine type II collagen in 100 μl of Freund complete adjuvant into the base of the tail.
Surgical destabilization of the medial meniscus (DMM) model of osteoarthritis was performed on 10-week-old C57BL/6 male mice, as described [23]. In brief, the right meniscotibial ligament was transected, resulting in the release of the medial meniscus from its tibial attachment.
In vivo localization of 1-11E/vIL-10 in AIA
1-11E/vIL-10 and the control C7/vIL-10 were labeled with Cy5.5 according to the manufacturer’s instructions (GE Healthcare, Buckinghamshire, UK), resulting in a dye-to-protein ratio of 2.2. Before injecting the tagged fusion protein into mice, its integrity was first assessed with ELISA and immunohistochemistry, as described earlier. To track the fusion proteins in vivo after inflammation, 1 μg of Cy5.5-labeled fusion protein was injected intraperitoneally (i.p.) 1 day after the mBSA re-challenge in the AIA model. Epifluorescence images were obtained daily after anesthesia induction with isofluorane in an IVIS Spectrum imager, by using an excitation wavelength of 675 nm and an emission wavelength of 720 nm (Perkin Elmer, Waltham, MA, USA). Images were analyzed by using Living Image 4.4 to obtain the average fluorescence intensities of a circular region of interest encompassing the knee joint. Four days after i./p. injection of Cy5.5-labeled fusion protein, a single animal was killed and the knees, liver, kidney, heart, and spleen imaged ex vivo. The knees were further embedded in optimal-cutting-temperature media (VWR, Leicestershire, UK), before freezing in isopentane cooled by liquid nitrogen, before storage overnight at -80°C. The joints were sectioned at a thickness of 15 μm by using a Leica CM1900UV cryotome (Leica Biosystems, Milton Keynes, UK). The cryosections were fixed in ice-cold acetone before being air dried and blocked in DAKO blocking solution (DAKO, Ely, Cambridgeshire, UK). Fluorescence confocal microscope images of the sections were obtained, as described earlier.
Treatment of AIA with 1-11E/vIL-10
On days 1 and 3 after the mBSA rechallenge in the AIA model, animals were injected i.p. with 30 μg of 1-11E/IL10 or C7/vIL-10. Swelling of the knee was measured daily by using calipers. On day 3, three animals were killed by cervical dislocation, serum was collected, and the knee joints were dissected and fixed in formalin (2% (vol/vol)) overnight, decalcified in EDTA for 5 weeks, and embedded in paraffin. Serial sections 2 μm in thickness were cut and further stained with safranin O. Serum cytokine concentrations were determined by using a seven-plex mouse pro-inflammatory assay kit (Mesoscale Discovery, Gaithersburg, MD, USA) in triplicate, according to the manufacturer’s instructions.
Levels of inflammation and redox state were determined by monitoring the luminescence after i.p. administration of 100 μl of 50 mg/ml luminol (5-amino-2,3-dihydro-1,4-phthalazine-dione; Sigma, Dorset, UK) in PBS. Luminol is a redox-sensitive compound that emits blue luminescence (λmax 425 nm) when exposed to ROS but depends on myeloperoxidase activity [17]. The luminescence was determined by analyzing images obtained 15 minutes after luminol injection in the IVIS Spectrum Bioluminescence settings.
Safranin O-stained knee sections were scored for disease as follows: 0, Control; 1, no subsynovial inflammation, synovial healthy chondrocytes; 2, some evidence of soft-tissue edema and subsynovial inflammation, some zones of partial-thickness loss of cartilage staining; 3, marrow involvement becoming apparent, frank soft-tissue edema, moderate inflammation with synovial thickening, inflammatory tissue encroaching into joint, nonadhered. Cartilage matrix depletion evident; no chondrocyte death; 4, Edema of soft tissue, subsynovial inflammation, pannus encroachment and adhesion to cartilage, full-thickness cartilage staining depletion, chondrocyte clum** and death, and inflamed marrow.
Statistical analysis
Statistical analyses were undertaken by using Prism (Graphpad, La Jolla, CA, USA) by using Newman-Keuls multiple-comparisons test. An α value of 0.05 was used as the threshold for significance.





