
Abstract
Background
Nilotinib is a second-generation tyrosine kinase inhibitor that exhibits significant efficacy as first- or second-line treatment in patients with chronic myeloid leukemia (CML). We conducted a multicenter Phase II Clinical Trial to evaluate the safety and efficacy of nilotinib among Japanese patients with imatinib-resistant or -intolerant CML-chronic phase (CP) or accelerated phase (AP).
Results
We analyzed 49 patients (33 imatinib-resistant and 16 imatinib-intolerant) treated with nilotinib 400 mg twice daily. The major molecular response (MMR) rate was 47.8% at 12 months among 35 patients who did not demonstrate an MMR at study entry. Somatic BCR-ABL1 mutations (Y253H, I418V, and exon 8/9 35-bp insertion [35INS]) were detected in 3 patients at 12 months or upon discontinuation of nilotinib. Although 75.5% of patients were still being treated at 12 months, nilotinib treatment was discontinued because of progressing disease in 1 patient, insufficient effect in 2, and adverse events in 9. There was no statistically significant correlation between MMR and trough concentrations of nilotinib. Similarly, no correlation was observed between trough concentrations and adverse events, except for pruritus and hypokalemia. Hyperbilirubinemia was frequently observed (all grades, 51.0%; grades 2–4, 29%; grades 3–4, 4.1%). Hyperbilirubinemia higher than grade 2 was significantly associated with the uridine diphosphate glucuronosyltransferase (UGT)1A9 I399C/C genotype (P = 0.0086; Odds Ratio, 21.2; 95% Confidence Interval 2.2–208.0).
Conclusions
Nilotinib was efficacious and well tolerated by patients with imatinib-resistant or -intolerant CML-CP/AP. Hyperbilirubinemia may be predicted before nilotinib treatment, and may be controlled by reducing the daily dose of nilotinib in patients with UGT1A9 polymorphisms.
Trial registration
clinicaltrials.gov: UMIN000002201
Similar content being viewed by others


Background
Imatinib is used as a first-line therapy for newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia (Ph + CML) [1, 2]; however, some patients fail to respond or become intolerant to this treatment [3]. Nilotinib is a second-generation tyrosine kinase inhibitor (2G-TKI) with higher selectivity and more potent inhibitory effects on the breakpoint cluster region-Abelson 1 (BCR-ABL1) tyrosine kinase than imatinib [4, 5]. Several studies have shown hematologic and cytogenetic responses to nilotinib in patients with imatinib-resistant or -intolerant CML [6–11]. Although point mutations in BCR-ABL1 are a major cause of imatinib-resistance, nilotinib is effective in patients with known point mutations in this oncogene, with the exception of the T315I mutation [3]. However, the frequency or profile of BCR-ABL1 point mutations has not been determined in daily practice when treating Japanese patients with imatinib-resistant CML in chronic phase (CP) or accelerated phase (AP).
Hyperbilirubinemia is one of the adverse events (AEs) caused by nilotinib. Recently, Giles et al. reported a correlation between hyperbilirubinemia and nilotinib trough concentrations in patients treated with nilotinib [12]. Moreover, nilotinib is not a substrate for uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) enzymes, but an inhibitor of human UGT1A1 in vitro [13], and CML patients with the UGT1A1*28 polymorphism show an increased risk of nilotinib-induced hyperbilirubinemia [14]. In Japanese cancer patients, UGT1A1*28, UGT1A9*1b, UGT1A1*6 (211G > A), and UGT1A1*60 (3279 T > G) are closely associated with the UGT1A9 IVS1 + 399 (I399C > T) polymorphism, and linkage of I399C > T with these variants has been shown to affect irinotecan metabolism [15]. However, we are not aware of any studies that have examined a correlation between hyperbilirubinemia and the UGT1A9 I399C > T polymorphism.
The goals of the present study were as follows: 1) to determine the major molecular response rate (MMR) at 12 months of twice daily (BID) treatment with 400 mg nilotinib in patients with imatinib-resistant/intolerant CML-CP or -AP; 2) to evaluate molecular responses associated with BCR-ABL1 mutation status or plasma concentrations of nilotinib; and 3) to evaluate the safety of administering 400 mg nilotinib BID, including hyperbilirubinemia development, based on plasma concentrations of nilotinib or polymorphisms of UGT1A1 and UGT1A9.
Results
Patients and treatment
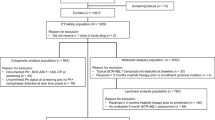
Between March 13, 2009 and January 12, 2011, 51 Japanese patients were recruited, and 49 patients (CML-CP, n = 45; CML-AP, n = 4) were included in the study. Two patients were excluded because they withdrew informed consent. The cut-off date for overall survival (OS) was January 11, 2013 (24 months after the last patient enrolled).
The demographic and baseline disease characteristics of the patients are described in Table 1. The median age was 62 years. The ratio of men to women was 27:22. Thirty-three (67.3%) and 16 (32.7%) patients were imatinib-resistant or imatinib-intolerant, respectively. The median duration of CML in patients was 46.0 months. The median duration of prior imatinib treatment was 43.4 months. Patients were treated with interferon (9 patients; 18.4%) with or without hydroxyurea (8 patients; 16.3%) prior to imatinib therapy. Twenty-three patients (46.9%) showed a complete cytogenetic response (CCyR), and 14 patients (28.6%) showed MMR at the time of entry into the study. Five patients (15.2%) had an additional chromosomal abnormality, and 6 patients (12.2%) had BCR-ABL1 kinase domain mutations at baseline.
Molecular response
Of the 49 patients on trial, 35 did not demonstrate an MMR at study entry and were evaluable for response, using the international scale of standardized quantitative real-time polymerase chain reaction (IS-PCR). The rates of MMR in the evaluable patients were 38.5% and 47.8% at 6 and 12 months, respectively, and the rates of MR4.5 were 7.7% and 13.0% at 6 and 12 months, respectively (Figure 1). The rates of BCR-ABL1 transcript levels (BCR-ABLIS) of <1% and <10% at 12 months were 74% and 87%, respectively.

Molecular responses in patients with imatinib-resistant and imatinib-intolerant CML treated with nilotinib. The molecular response was evaluated according to the international scale of standardized quantitative real-time polymerase chain reaction at the beginning of the study and every 3 months thereafter. The X-axis shows the time-point and patient number for molecular response evaluation. The Y-axis shows the percentage of patients who achieved a molecular response.
The cumulative MMR rates at 12 months were 62.5%, 33.3%, and 24.8% in patients with baselines >0.1–1%, >1–10%, and >10%, respectively. These data represented a significant difference between patients with baseline >0.1–1% and those with baseline >10% (log-rank test, P = 0.0372). The median BCR-ABL1 transcript levels (BCR-ABLIS) reduced significantly from 10.71% to 0.14% by 12 months in a time dependent manner (Figure 2).

Kinetics of BCR-ABL1 expression. The black bars indicate the median. The gray bars indicate quartiles 1 and 3. The X-axis shows the time-point and patient number for evaluation of molecular responses. The Y-axis shows BCR-ABL1 transcript levels per the international scale of standardized quantitative real-time polymerase chain reaction.
BCR-ABL1 mutations and nilotinib trough concentrations
Using the direct sequencing identification method (DS), 5 BCR-ABL1 mutations (M244V, F317L, N358D, F359V, and E459K) were detected in 6 patients (12.2%) at baseline. However, M244V, N358D, and E459K were undetectable after nilotinib treatment at 12 months. In contrast, new somatic BCR-ABL1 mutations (Y253H, I418V, and exon 8/9 35-bp insertion [35INS]) were detected in 3 patients (6.1%) at 12 months. These 3 patients did not achieve MMR. The somatic BCR-ABL1 mutation (T315I) was not detected.
Nilotinib trough plasma concentrations were measured by high-performance liquid chromatography (HPLC) methods using blood collected at 3 months. Among 20 patients who continued treatment with nilotinib for 12 months and did not show somatic BCR-ABL1 mutations, trough concentrations (median, 25% to 75%) were increased in patients who achieved MMR at 12 months (774.4 ng/mL, 584.0 to 1364.0) compared to patients without MMR (489.7 ng/mL, 317.6 to 922.5). However, the correlation between MMR at 12 months and trough concentrations of nilotinib was not statistically significant (P = 0.261).
Patient status
The disposition of patients at 12 months is summarized in Table 2. Thirty-seven patients (75.5%) were still being treated with nilotinib treatment, while treatment was discontinued for 12 patients (24.5%). AEs were the most frequent reasons for terminating treatment (9 patients; 18.4%). CML-CP progressed in 1 patient with an F317L mutation at baseline. The median actual dose intensity (total nilotinib dose/treatment time) was 651 mg/day. The estimated 36-month OS was 95% in the 37 patients on treatment and 60% in the 12 patients for which treatment was terminated. These data represented a significant difference between the two groups (Figure 3; log-rank test, P = 0.00844).

Overall survival of patients on present or discontinued treatment after 12 months. OS was measured from the date of initial nilotinib administration to the date of death, with a minimum follow-up of 24 months. OS was estimated using the Kaplan-Meier method and compared between groups using the stratified log-rank test. There was a significant difference between the 2 groups (log-rank test, P = 0.00844).
Safety
Nilotinib therapy was well tolerated. Although severe nonhematologic AEs were infrequent, 1 patient presenting with grade 3/4 headache and 1 with ischemic heart disease discontinued nilotinib therapy. Grade 3/4 hematologic AEs included anemia (14.3%), neutropenia (28.6%), and thrombocytopenia (18.4%). Although hematologic AEs were managed with dose interruptions or reductions, 5 patients discontinued nilotinib because of repeated or prolonged grade 3/4 thrombocytopenia. Although corrected QT interval (QTc) prolongation at all grades was found in only three patients (6.1%), grade 3/4 QTc prolongation was not detected. QTc prolongation was also easily managed with dose interruptions or reductions, and no patients discontinued treatment because of this AE. Moreover, no episodes of torsades de pointes or death due to arrhythmias were noted. However, 1 patient discontinued nilotinib therapy because of ischemic heart disease without QTc prolongation.
Grade 3/4 elevations of total bilirubin occurred in 2 patients (4.1%), and their nilotinib therapy was discontinued because of repeated hyperbilirubinemia despite dose interruptions or reductions. Although other biochemical abnormalities were generally mild, transient, and easily managed with dose interruptions or reductions, hyperbilirubinemia was frequently observed (all grades, 51.0%; grades 2–4, 29%). There was no statistically significant correlation between AEs and trough concentrations of nilotinib except for pruritus (P = 0.0440) and hypokalemia (P = 0.0221).
The UGT1A1 and UGT1A9 polymorphisms were analyzed in all patients to identify a correlation with laboratory abnormalities related to liver function. Although we did not observe a significant correlation between hyperbilirubinemia and UGT1A1 genotype (poor metabolizers with UGT1A1*6/*28, *27/*28, or *28/*28 vs. extensive metabolizers with UGT1A1 *1/*1, *1/*6, *1/*27, or *1/*28), there was a significant correlation with the UGT1A9 I399C/C genotype (P = 0.028) (Table 3). This genotype was observed in 33% of patients with hyperbilirubinemia higher than grade 2. The UGT1A9 I399C/C genotype was a statistically independent factor according to the results of stepwise forward selection multiple regression analysis (P = 0.0086, Odds Ratio, 21.2; 95% Confidence Interval, 2.2–208.0).
Discussion
The present study revealed that nilotinib therapy was effective in Japanese patients with CML-CP/AP who have developed imatinib resistance or intolerance. Approximately 50% of the patients in this Phase II trial achieved MMR, and the estimated OS at 36 months for patients on treatment was 95%. Responses were more likely in patients with BCR-ABLIS ≤1% at baseline than in those with BCR-ABLIS >1%. In a phase II trial of nilotinib for imatinib resistance or intolerance reported by Kantarjian et al. [9], MMR was achieved in 32% (43 of 134) of patients at 12 months. The lower MMR reported in that study as compared to the rate observed in the present study might be explained by differences in response at baseline. The CCyR at baseline observed by Kantarjian et al. [9] was 3%; in comparison, 26% (9 of 35 patients) showed this response in our trial. In the Expanding Nilotinib Access in Clinical Trials (ENACT), the molecular responses in a subset of French patients (n = 168) every 3 months were evaluated. Thirty-seven percent achieved MMR, and 20% achieved MR4.5 by 12 months [10]. In a phase I/II study of nilotinib treatment of Japanese patients with imatinib-resistant or -intolerant CML or relapsed/refractory Ph + acute lymphoblastic leukemia (ALL), 16 patients with CML-CP were assessed using IS-PCR to detect molecular responses every 3 months [6]. The level of BCR-ABL transcription gradually decreased from baseline, with a 1-log reduction at 6 months and a 2-log reduction at 12 months. Our findings are consistent with these observations and suggest that nilotinib is highly active in patients with imatinib-resistant or -intolerant CML-CP/AP.
We detected 5 BCR-ABL mutations (M244V, F317L, N358D, F359V, and E459K) in 6 patients at baseline screening. Among these patients, M244V, N358D, and E459K were not detected after nilotinib treatment at 12 months, and 3 patients with these mutations achieved MMR. In contrast, patients with F317L and F359V + E459K mutations showed poor responses to nilotinib treatment, and 1 patient with F317L experienced disease progression. The remaining patient with F359V + E459K mutations did not respond to nilotinib, and treatment was discontinued because of grade 3 thrombocytopenia. A previous study showed that patients with F317L or F359V mutations are resistant to nilotinib [16, 17]. The present study also suggests that these mutations at baseline may be associated with less favorable responses to nilotinib.
We detected 3 new mutations in 3 patients after they started nilotinib treatment (Y253H, I418V, and exon 8/9 35-bp insertion). Although the Y253H mutation was less sensitive to nilotinib in an in vitro cell viability assay (IC50 > 150 nM) [17], the level of BCR-ABL1 transcripts in the Y253H patient in our trial decreased from 8.06% to 0.30%. Qin et al. [18] reported the I418V mutation in a Chinese patient who progressed to blast phase while on imatinib therapy. Although a patient with the somatic I418V mutation in our trial was in second chronic phase at baseline, the disease did not progress. However, the levels of the BCR-ABL1 transcript were maintained from 59.36% at baseline to 50.21% at 12 months. Another rare mutation of BCR-ABL1 is 35INS, which is a 35-bp insertion between ABL kinase domain exons 8 and 9 [19]. This insertion results in a frameshift, leading to the addition of 10 residues and truncation of 653C-terminal residues due to early termination [20]. One patient with 35INS in our trial showed a mild reduction in the level of BCR-ABL1 transcripts from 10.71% at baseline to 2.89% at 12 months. O’Hare et al. reported that 35INS is kinase-inactive and does not contribute to TKI resistance [21]. Patients who did not achieve MMR and harbored somatic mutations showed relatively poor responses to nilotinib. However, a mutation (e.g. 35INS) might not be causally related, suggesting the possibility of genomic instability, and that other genetic abnormalities might contribute to TKI resistance. Conversely, the T315I mutation, which is known to cause resistance to imatinib and nilotinib, was not detected in the present study.
In the Evaluating Nilotinib Efficacy and Safety in Clinical Trials-newly diagnosed (ENESTnd) patients, the occurrence of all-grade total bilirubin elevation was significantly higher in patients with higher nilotinib exposure [22]. Although we could not identify a correlation between nilotinib trough concentrations and hyperbilirubinemia in the present study, nilotinib trough concentration has been associated with the occurrence of all-grade elevations in total bilirubin [12]. However, to our knowledge this is the first report of a significant correlation between hyperbilirubinemia and the UGT1A9 I399C/C genotype.
UGT1A1 catalyzes gluconidation of hepatic bilirubin in humans [23]. The presence of 7-TA nucleotide repeats in the (TA)nTAA promoter region of UGT1A1 (UGT1A1*28) leads to decreased expression of this gene, resulting in high plasma bilirubin levels that form the basis for Gilbert’s syndrome [24]. Because nilotinib is a potent noncompetitive inhibitor of human UGT1A1 activity [13], the UGT1A1*28 polymorphism increases the risk of nilotinib induced-hyperbilirubinemia [14, 25]. These observations suggest that nilotinib-associated hyperbilirubinemia is very likely the result of inhibition of UGT1A1 activity, combined with genetic defects in UGT1A1. Although, UGT1A9 is also an enzyme of the glucuronidation pathway that transforms small lipophilic molecules, such as steroids, bilirubin, hormones, and drugs, into water-soluble, excretable metabolites, we are not aware of any reports indicating that nilotinib-induced hyperbilirubinemia is related to the UGT1A9 I399C/C genotype. It has been shown that the enzyme function of the UGT1A9 I399C > T genotype does not contribute to variability in UGT1A9 activity [26]. On the other hand, Saito et al. reported the role of UGT1A9 I399C > T in SN-38 glucuronidation in a study of 177 Japanese cancer patients administered irinotecan. Haplotype analysis showed that 98% of UGT1A9 I399C alleles was linked to low-activity genotypes, either UGT1A1*6, *28, or *60 [15]. Our study suggests that hyperbilirubinemia is associated with the UGT1A9 I399C/C genotype. For Japanese patients with CML, hyperbilirubinemia may be predicted before nilotinib treatment and controlled by reducing the daily dose of nilotinib in patients with UGT1A9 polymorphisms.
Conclusions
The results of our study suggest that nilotinib is generally well tolerated by patients and effectively treats imatinib-resistant or -intolerant CML-CP/AP.
Methods
Eligibility criteria
Twenty-seven institutions in the East Japan CML study group (EJCML) participated. Patients with Ph + CML-CP/AP who were at least 18 years of age were eligible if they were imatinib-resistant or -intolerant, demonstrated adequate performance status (World Health Organization [WHO] Performance Score [PS] <2), and had normal hepatic, renal, and cardiac functions. Patients were excluded if they exhibited any of the following characteristics: blast phase, QTc >450 ms, and the T315I mutation. Imatinib resistance was defined as an incomplete hematologic response at or after 3 months, no MCyR at 6 months, no CCyR at 12 months, or no MMR at 18 months, according to the criteria of failure and suboptimal response of European LeukemiaNet [3]. Imatinib intolerance was defined as a lack of optimal response because of a grade 3/4 imatinib-related AE, or because of a persistent grade 2 imatinib-related AE, despite optimal supportive care, that persisted for more than 1 month or recurred more than 3 times following reduction of the dose of imatinib.
Clinical trial design and objectives
The study was conducted in accordance with the principles of the Declaration of Helsinki. Informed consent was written by all patients according to institutional guidelines. The study was approved by all institutional review boards and registered with ClinicalTrials.gov (number UMIN000002201). The primary objective of this phase II, single-treatment arm, open-label study was to determine the incidence of MMR at 12 months in patients with imatinib-resistant/intolerant CML-CP/AP who were treated with nilotinib. The molecular response was evaluated according to IS-PCR upon study entry and every 3 months thereafter. Secondary objectives were to evaluate the relationship between the molecular response and BCR-ABL1 point mutations or plasma concentrations of nilotinib, and between the safety profile of nilotinib and plasma concentrations of the drug and or UGT 1A1/1A9 polymorphisms.
Dosing regimen and concomitant therapy
Nilotinib was administered orally at 400 mg BID, at approximately 12-h intervals. Food was not consumed with the drug, or for at least 2 h before and 1 h after the drug was ingested. The concomitant administration of a strong inhibitor, including grapefruit juice, or inducer of CYP3A, was not allowed in the study.
Molecular response
IS-PCR was performed in a central laboratory (BML Inc., Kawagoe, Japan) using the MolecularMD One-Step qRT-PCR BCR-ABL kit (MolecularMD, Cambridge, MA). An MMR was defined as a 3-log reduction in the level of the BCR-ABL1 transcript using IS-PCR (BCR-ABLIS 0.0032–0.1%). At least 10,000 control genes (ABL1) were required for a sample to be classified as adequate. MR4.5 was defined as ≥4.5-log reduction in the levels of the BCR-ABL1 transcript (BCR-ABLIS ≤0.0032%) in peripheral blood samples, and at least 32,000 control genes were required for a sample to be classified as adequate. Patients with MMR at baseline were considered not eligible and were excluded from the analysis of response rates.
Measurement of nilotinib concentration and Genoty**
Nilotinib trough concentrations (C0) were determined using HPLC as described previously [27]. BCR-ABL1 mutations were analyzed by the DS method as previously described [28]. Genoty** of UGT1A1*6, *27 and *28 was performed using PCR-restriction fragment length polymorphism as described [29, 30]. The analysis results obtained from PCR-RFLP were confirmed using a fully automated single nucleotide polymorphism (SNP) detection system (prototype i-densy™, ARKRAY Inc., Kyoto, Japan). The UGT1A9 I399C > T (relative to the end of UGT1A9 exon 1) polymorphism was genotyped by direct sequencing using a PCR procedure as previously described [31]. All genotype frequencies were tested for their consistency with Hardy-Weinberg equilibrium.
Safety analyses
Safety was assessed by determining the frequency and severity of AEs, which included hematologic and biochemical laboratory tests, vital signs, physical examinations (including body weight), WHO PS cardiac function tests (12-lead electrocardiogram, cardiac enzyme test, and echocardiography), and chest X-rays. AEs were graded according to the National Cancer Institute Common Terminology Criteria for AEs (version 3.0).
Statistical analyses
Statistical analyses were performed using SPSS statistical software (version 17.0, SPSS Japan Inc., Tokyo, Japan). Data were presented as number or median values (range or 25% to 75%). Differences in the various parameters between groups were evaluated using the Mann–Whitney U or chi-square tests. Time to MMR was measured from the date of initial nilotinib administration to the date of MMR during the study. OS was measured from the date of initial nilotinib administration to the date of death with a minimum follow-up of 24 months. OS and the cumulative incidence of MMR were estimated using the Kaplan-Meier method and compared between groups using the stratified log-rank test. Stepwise forward selection multiple regression analysis was performed to determine the effect of the variables examined in univariate analysis. A P value of <0.05 was considered significant.
Abbreviations
- CML:
-
Chronic myeloid leukemia
- Ph:
-
Philadelphia chromosome
- CP:
-
Chronic phase
- AP:
-
Accelerated phase
- UGT:
-
Uridine diphosphate glucuronosyltransferase
- MMR:
-
Major molecular response
- 2G-TKI:
-
Second-generation tyrosine kinase inhibitor
- BCR-ABL1:
-
Breakpoint cluster region-Abelson 1
- AEs:
-
Adverse events
- OS:
-
Overall survival
- CCyR:
-
Complete cytogenetic response
- MCyR:
-
Major cytogenetic response
- IS-PCR:
-
The international scale of standardized quantitative real-time polymerase chain reaction
- DS:
-
The direct sequencing identification method
- 35INS:
-
Exon8/9 35-bp insertion
- QTc:
-
Corrected QT interval
- HPLC:
-
High-performance liquid chromatography
- WHO:
-
World Health Organization
- PS:
-
Performance score.
References
O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A, Goldman JM, Kantarjian H, Taylor K, Verhoef G, Bolton AE, Capdeville R, Druker BJ, IRIS Investigators: Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003, 348: 994–1004. 10.1056/NEJMoa022457
O’Brien SG, Guilhot F, Goldman JM, Hochhaus A, Hughes TP, Radich JP, Rudoltz M, Filian J, Gathmann I, Druker BJ, Larson RA: International Randomized Study of Interferon Versus STI571 (IRIS) 7-Year Follow-up: Sustained Survival, Low Rate of Transformation and Increased Rate of Major Molecular Response (MMR) in Patients (pts) with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CMLCP) Treated with Imatinib (IM) [abstract]. Blood 2008, 112: 186.
Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, Cervantes F, Deininger M, Gratwohl A, Guilhot F, Hochhaus A, Horowitz M, Hughes T, Kantarjian H, Larson R, Radich J, Simonsson B, Silver RT, Goldman J, Hehlmann R, European LeukemiaNet: Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009, 27: 6041–6051. 10.1200/JCO.2009.25.0779
Kantarjian HM, Hochhaus A, Saglio G, De Souza C, Flinn IW, Stenke L, Goh YT, Rosti G, Nakamae H, Gallagher NJ, Hoenekopp A, Blakesley RE, Larson RA, Hughes TP: Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol 2011, 12: 841–851. 10.1016/S1470-2045(11)70201-7
Kantarjian HM, Kim DW, Issaragrisil S, Clark RE, Reiffers J, Nicolini FE, Pasquini R, Kalaycio M, Hughes TP, Hochhaus A, Kemp C, Fan X, Waltzman RJ, Saglio G, Larson RA: Enestnd 4-year update: continued superiority of Nilotinib vs Imatinib in patients with newly diagnosed Philadelphia chromosome–positive chronic myeloid leukemia in chronic phase [abstract]. Blood 2012, 120: 1676.
Usuki K, Tojo A, Maeda Y, Kobayashi Y, Matsuda A, Ohyashiki K, Nakaseko C, Kawaguchi T, Tanaka H, Miyamura K, Miyazaki Y, Okamoto S, Oritani K, Okada M, Usui N, Nagai T, Amagasaki T, Wanajo A, Naoe T: Efficacy and safety of nilotinib in Japanese patients with imatinib-resistant or -intolerant Ph + CML or relapsed/refractory Ph + ALL: a 36-month analysis of a phase I and II study. Int J Hematol 2012, 95: 409–419. 10.1007/s12185-012-1026-9
Tojo A, Usuki K, Urabe A, Maeda Y, Kobayashi Y, **nai I, Ohyashiki K, Nishimura M, Kawaguchi T, Tanaka H, Miyamura K, Miyazaki Y, Hughes T, Branford S, Okamoto S, Ishikawa J, Okada M, Usui N, Tanii H, Amagasaki T, Natori H, Naoe T: A Phase I/II study of nilotinib in Japanese patients with imatinib-resistant or -intolerant Ph + CML or relapsed/refractory Ph + ALL. Int J Hematol 2009, 89: 679–688. 10.1007/s12185-009-0327-0
Giles FJ, Le Coutre PD, Pinilla-Ibarz J, Larson RA, Gattermann N, Ottmann OG, Hochhaus A, Radich JP, Saglio G, Hughes TP, Martinelli G, Kim DW, Novick S, Gillis K, Fan X, Cortes J, Baccarani M, Kantarjian HM: Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013, 27: 107–112. 10.1038/leu.2012.181
Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz J, Larson RA, Gattermann N, Ottmann OG, Hochhaus A, Radich JP, Saglio G, Hughes TP, Martinelli G, Kim DW, Shou Y, Gallagher NJ, Blakesley R, Baccarani M, Cortes J, Le Coutre PD: Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood 2011, 117: 1141–1145. 10.1182/blood-2010-03-277152
Nicolini FE, Turkina A, Shen ZX, Gallagher N, Jootar S, Powell BL, De Souza C, Zheng M, Szczudlo T, Le Coutre P: Expanding Nilotinib Access in Clinical Trials (ENACT): an open-label, multicenter study of oral nilotinib in adult patients with imatinib-resistant or imatinib-intolerant Philadelphia chromosome-positive chronic myeloid leukemia in the chronic phase. Cancer 2012, 118: 118–126. 10.1002/cncr.26249
Branford S, Kim DW, Soverini S, Haque A, Shou Y, Woodman RC, Kantarjian HM, Martinelli G, Radich JP, Saglio G, Hochhaus A, Hughes TP, Müller MC: Initial molecular response at 3 months may predict both response and event-free survival at 24 months in imatinib-resistant or -intolerant patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase treated with nilotinib. J Clin Oncol 2012, 30: 4323–4329. 10.1200/JCO.2011.40.5217
Giles FJ, Yin OQ, Sallas WM, Le Coutre PD, Woodman RC, Ottmann OG, Baccarani M, Kantarjian HM: Nilotinib population pharmacokinetics and exposure-response analysis in patients with imatinib-resistant or -intolerant chronic myeloid leukemia. Eur J Clin Pharmacol 2013, 69: 813–823. 10.1007/s00228-012-1385-4
Fujita K, Sugiyama M, Akiyama Y, Ando Y, Sasaki Y: The small-molecule tyrosine kinase inhibitor nilotinib is a potent noncompetitive inhibitor of the SN-38 glucuronidation by human UGT1A1. Cancer Chemother Pharmacol 2011, 67: 237–241. 10.1007/s00280-010-1445-3
Singer JB, Shou Y, Giles F, Kantarjian HM, Hsu Y, Robeva AS, Rae P, Weitzman A, Meyer JM, Dugan M, Ottmann OG: UGT1A1 promoter polymorphism increases risk of nilotinib-induced hyperbilirubinemia. Leukemia 2007, 21: 2311–2315. 10.1038/sj.leu.2404827
Saito Y, Sai K, Maekawa K, Kaniwa N, Shirao K, Hamaguchi T, Yamamoto N, Kunitoh H, Ohe Y, Yamada Y, Tamura T, Yoshida T, Minami H, Ohtsu A, Matsumura Y, Saijo N, Sawada J: Close association of UGT1A9 IVS1 + 399C > T with UGT1A1*28, *6, or *60 haplotype and its apparent influence on 7-ethyl-10-hydroxycamptothecin (SN-38) glucuronidation in Japanese. Drug Metab Dispos 2009, 37: 272–276. 10.1124/dmd.108.024208
Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, Gambacorti-Passerini C, Boschelli F: Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol 2009, 27: 469–471.
Hughes T, Saglio G, Branford S, Soverini S, Kim DW, Muller MC, Martinelli G, Cortes J, Beppu L, Gottardi E, Kim D, Erben P, Shou Y, Haque A, Gallagher N, Radich J, Hochhaus A: Impact of baseline BCR-ABL mutations on response to nilotinib in patients with chronic myeloid leukemia in chronic phase. J Clin Oncol 2009, 27: 4204–4210. 10.1200/JCO.2009.21.8230
Qin Y, Chen S, Jiang B, Jiang Q, Jiang H, Li J, Li L, Lai Y, Liu Y, Huang X: Characteristics of BCR-ABL kinase domain point mutations in Chinese imatinib-resistant chronic myeloid leukemia patients. Ann Hematol 2011, 90: 47–52. 10.1007/s00277-010-1039-5
Laudadio J, Deininger MW, Mauro MJ, Druker BJ, Press RD: An intron-derived insertion/truncation mutation in the BCR-ABL kinase domain in chronic myeloid leukemia patients undergoing kinase inhibitor therapy. J Mol Diagn 2008, 10: 177–180. 10.2353/jmoldx.2008.070128
Lee TS, Ma W, Zhang X, Giles F, Cortes J, Kantarjian H, Albitar M: BCR-ABL alternative splicing as a common mechanism for imatinib resistance: evidence from molecular dynamics simulations. Mol Cancer Ther 2008, 7: 3834–3841. 10.1158/1535-7163.MCT-08-0482
O’Hare T, Zabriskie MS, Eide CA, Agarwal A, Adrian LT, You H, Corbin AS, Yang F, Press RD, Rivera VM, Toplin J, Wong S, Deininger MW, Druker BJ: The BCR-ABL35INS insertion/truncation mutant is kinase-inactive and does not contribute to tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Blood 2011, 118: 5250–5254. 10.1182/blood-2011-05-349191
Larson RA, Yin OQ, Hochhaus A, Saglio G, Clark RE, Nakamae H, Gallagher NJ, Demirhan E, Hughes TP, Kantarjian HM, Le Coutre PD: Population pharmacokinetic and exposure-response analysis of nilotinib in patients with newly diagnosed Ph + chronic myeloid leukemia in chronic phase. Eur J Clin Pharmacol 2012, 68: 723–733. 10.1007/s00228-011-1200-7
Bosma PJ, Seppen J, Goldhoorn B, Bakker C, Oude Elferink RP, Chowdhury JR, Chowdhury NR, Jansen PL: Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem 1994, 269: 17960–17964.
Strassburg CP: Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome). Best Pract Res Clin Gastroenterol 2010, 24: 555–571. 10.1016/j.bpg.2010.07.007
Shibata T, Minami Y, Mitsuma A, Morita S, Inada-Inoue M, Oguri T, Shimokata T, Sugishita M, Naoe T, Ando Y: Association between severe toxicity of nilotinib and UGT1A1 polymorphisms in Japanese patients with chronic myelogenous leukemia. Int J Clin Oncol 2013. [Epub ahead of print]
Ramirez J, Liu W, Mirkov S, Desai AA, Chen P, Das S, Innocenti F, Ratain MJ: Lack of association between common polymorphisms in UGT1A9 and gene expression and activity. Drug Metab Dispos 2007, 35: 2149–2153. 10.1124/dmd.107.015446
Miura M, Takahashi N, Sawada K: High-performance liquid chromatography with solid-phase extraction for the quantitative determination of nilotinib in human plasma. Biomed Chromatogr 2010, 24: 789–793.
Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP: High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002, 99: 3472–3475. 10.1182/blood.V99.9.3472
Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y: Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 2000, 60: 6921–6926.
Miura M, Kagaya H, Satoh S, Inoue K, Saito M, Habuchi T, Suzuki T: Influence of drug transporters and UGT polymorphisms on pharmacokinetics of phenolic glucuronide metabolite of mycophenolic acid in Japanese renal transplant recipients. Ther Drug Monit 2008, 30: 559–564. 10.1097/FTD.0b013e3181838063
Girard H, Villeneuve L, Court MH, Fortier LC, Caron P, Hao Q, Von Moltke LL, Greenblatt DJ, Guillemette C: The novel UGT1A9 intronic I399 polymorphism appears as a predictor of 7-ethyl-10-hydroxycamptothecin glucuronidation levels in the liver. Drug Metab Dispos 2006, 34: 1220–1228. 10.1124/dmd.106.009787
Acknowledgments
The authors thank the East Japan CML study group (EJCML), Ibaraki Hematology, Oncology and Palliation Expert Meeting (IB-HOPE), and the Akita CML study group for their participation in this study. This work was supported by a grant (No. 23590168) from the Japan Society for the Promotion of Science, Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
NT, KM, TK, YK, CY, SS, HH and KS receive research funding and honoraria for lectures from Novartis Pharmaceuticals, and Bristol-Myers Squibb. The other authors declare that they have no competing interests.
Authors’ contributions
NT and KS designed and performed the research. NT wrote the paper. TN, MA, and MM performed the pharmacokinetic and pharmacogenetic analyses. NT, TN, and MM analyzed the data and prepared the figures. JK, KM, AK, OS, HK, KI, NT, HN, TK, MM, YK, KT, MM, HS, CY, YT, TK, YW, JN, SY, KM, RH, TS, SS, AS, and HH participated in the study design and data analysis. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Takahashi, N., Miura, M., Kuroki, J. et al. Multicenter phase II clinical trial of nilotinib for patients with imatinib-resistant or -intolerant chronic myeloid leukemia from the East Japan CML study group evaluation of molecular response and the efficacy and safety of nilotinib. Biomark Res 2, 6 (2014). https://doi.org/10.1186/2050-7771-2-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2050-7771-2-6