
Abstract
Background
Plant cell walls are complex structures that full-fill many diverse functions during plant growth and development. It is therefore not surprising that thousands of gene products are involved in cell wall synthesis and maintenance. However, functional association for the majority of these gene products remains obscure. One useful approach to infer biological associations is via transcriptional coordination, or co-expression of genes. This approach has proved useful for several biological processes. Nevertheless, combining co-expression with other large-scale measurements may improve the biological inferences.
Results
In this study, we used a combined approach of co-expression and cell wall metabolomics to obtain new insight into cell wall synthesis in rice. We initially created a weighted gene co-expression network from publicly available datasets, and then established a comprehensive cell wall dataset by determining cell wall compositions from 29 tissues that almost cover the whole life cycle of rice. We subsequently combined the datasets through the conversion of co-expressed gene modules into eigen-vectors, representing expression profiles for the genes in the modules, and performed comparative analyses against the cell wall contents. Here, we made three major discoveries. First, we confirmed our approach by finding primary and secondary wall cellulose biosynthesis modules, respectively. Second, we found co-expressed modules that strongly correlated with re-organization of the secondary cell walls and with modifications and degradation of hemicellulosic structures. Third, we inferred that at least one module is likely to play a regulatory role in the production of G-rich lignification.
Conclusions
Here, we integrated transcriptomic associations and cell wall metabolism and found that certain co-expressed gene modules are positively correlated with distinct cell wall characteristics. We propose that combining multiple data-types, such as coordinated transcription and cell wall analyses, may be a useful approach to glean new insight into biological processes. The combination of multiple datasets, as illustrated here, can further improve the functional inferences that typically are generated via a single type of datasets. In addition, our data extend the typical co-expression approach to allow deeper insight into cell wall biology in rice.
Similar content being viewed by others


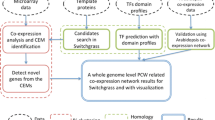
Background
Gene co-expression analyses can reveal functional relationships between gene products. These types of relationships are typically explored using some type of similarity measure, e.g. Pearson’s correlation coefficient (PCC), to quantify the association between two genes in the genome. The pairwise relationships can be represented as a network structure, in which edges (co-expression relationships) connect nodes (genes) that generally include the majority of genes in a given organism’s genome [1]. Based on these associations, it is possible to predict functional gene clusters, or groups of genes, that participate in common biological pathways [2, 3]. Moreover, this approach may also be used to find the conserved orthologous gene clusters across several species [4, 5], with the implication that the clusters are involved in similar biological processes.
Many co-expression networks have been constructed in plants, such as Arabidopsis [1, 3, 6–11], barley [12], rice [13, 14], poplar [15], tobacco [16], and maize [17]. Several of these efforts have been implemented as web-based tools, e.g. the Arabidopsis Co-expression Toolkit (ACT) [18], ATTED-II [19], AtCOECis [20], RiceArrayNet (PlantArraynet) [14], Co-expressed biological processes (CoP) database [15], The Gene Co-expression Network Browser [13], and two AraNets [1, 9], and PlaNet [21].
While the co-expression-based approaches have proved successful for several biological processes, far from all cellular aspects can rely on this type of metrics. Instead, integrative approaches are increasingly applied to extend knowledge gleaned from one type of dataset. These studies are typically relying on functional and structural genomics data, such as high-throughput microarray assays, next-generation sequencing, and metabolomic and proteomic technologies [22].
Plant cell walls are mainly composed of cellulose, non-cellulosic polysaccharides (hemicelluloses and pectin) and lignin, and represent the most abundant renewable biomass on earth [23]. The primary and secondary cell walls are typically distinct structures in plant growth and development [23], where the primary wall is a flexible matrix that allows directed cell growth and the secondary wall is a robust structure surrounding cells that need extra support for their functions. In general, cellulose makes up almost 25-30% of dry matter in grasses [24] and 40-45% in woody plants [25]. Hemicelluloses are polysaccharides that contain xyloglucans, xylans, mannans and glucomannans, and β-(1 → 3,1 → 4)-glucans, whereas pectins are diversified compounds that mainly are present in primary walls [26]. Lastly, the polyphenolic molecule lignin is an amorphous polymer of phenylpropane units with three monomers: p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) [27, 28], laid down during secondary wall formation.
More than one thousand gene products have been proposed to be dedicated to plant cell wall biogenesis and modification [29–1), we summarized the information using R function collapseRows[48, 49]. The resulting expression matrix contained 33,204 genes or probe sets. To be able to statistically compare the expression matrix to the cell wall data, we decided to construct a weighted correlation network [50] based on the 33,204 probes for the 29 tissues that we also used for cell wall analyses. Here, the weights of edges in the corresponding co-expression network correspond to the degree of similarity of the expression profiles of two adjacent nodes/genes.
Subsequently, a clustering approach of the weighted correlation network was undertaken, which resulted in 56 groups of highly co-expressed genes, also referred to as gene modules (Additional file 2). Hence, modules were defined as groups of genes which exhibit a high intra-module topological overlap [51]. The modules were denoted by numbers from zero to 55 and prefixed with “ME” referring to “module eigengene” [50]. Obviously, the numbers of genes (probe sets) per module differed, and more than half of the modules contained less than 500 genes (probe sets) (Additional file 3A). To explore the co-expression relationships between modules, a module’s representative expression pattern was summarized using the first principal component of all the module’s gene members. Further, all module eigengenes were clustered by using complete linkage method (Additional file 3B), which characterizes the similarity structure between the modules.
Biological relevance and connectivity scores of network modules
To assess the functional relevance of the gene modules, and to make sure that the co-expressed modules reflect biologically relevant information, we next examined whether certain ontology terms were over-represented in the modules. Gene ontology (GO) enrichment analysis was therefore performed using a weighted method and Fisher’s exact test [52] (Additional file 4). The analysis showed that a total of 4,014 enriched terms and 1,175 unique terms were identified among the modules at p < 0.05. Notably, a significant over-representation of the terms cellulose and non-cellulosic polysaccharide biosynthesis was observed for Module 24 (with 406 genes or probe sets) and Module 44 (with 136 genes or probe sets) (Additional file 4). Based on the representation of KEGG reference pathway maps and BRITE functional hierarchies [53], we furthermore performed a functional enrichment analysis of KEGG Orthology for each module using hypergeometric tests. Module 24 and Module 44 were enriched in glycan biosynthesis and metabolism, consistent with the findings that genes in Module 24 and Module 44 may participate in cellulose and non-cellulosic biosynthesis as observed in the GO enrichment analysis. Detailed significant associations for each module are supplied in Additional file 5.
Highly connected genes, or hubs, are thought to play a central role in biological networks. Connectivity has been found as an important complementary gene screening variable for finding biologically significant genes in particular biological processes [54]. Intramodular connectivity (kWithin) is defined as the gene connectivity inside a given module. In weighted networks, intramodular connectivity equals the sum of connection weights of a node with all other nodes inside module. In this study, we defined whole network connectivity kTotal, and external module connectivity (kOut = kTotal-kWithin) for any given node. To find genes of high connectivity (i.e. 'hubs’) in consensus modules, we evaluated the module eigengene-based connectivity (kME) as the correlation between the gene expression and the module eigengene [55]. We also calculated all connectivity types in all models, and the genes sorted out by their kME were listed in Additional file 6.
Cell wall composition analysis
In an attempt to assign certain cell wall related functions to the modules, we harvested material from the 29 tissues that corresponded to the microarray data sets above. We sequentially extracted wall polysaccharides including pectin with ammonium oxalate, hemicelluloses with KOH, and cellulose in the remaining pellet [56, 57]. The pectin was present at very low levels, or absent, in most rice tissues, and we therefore did not use the pectin data for any further investigation in this work. In summary, the cell wall composition varied greatly across the 29 tissues (Figure 1; Additional file 7). Cellulose content ranged from 0.29% (endosperm1) to 31.33% of dry matter (palea/lemma) (Figure 1A). Three major monosaccharides of hemicelluloses also varied significantly [60, 81] was used to align the nucleotide sequences of the remaining probe sets to the Michigan State University (MSU) Rice Genome Annotation version 6.0 [82] which currently contains 56,797 protein-coding gene models (BLAT parameters used: minIdentity = 100, minMatch = 1, stepSize = 5). Subsequently, 31,574 probe sets could be mapped to a unique genomic location with at least six perfect match probes (more than 50% of the 11 spotted probe-pairs per sequence). The probe sets in the expression matrix were annotated with the corresponding genes names; probe sets which could not be mapped to genes remain annotated with their original probe names. Further, to obtain a single expression level estimate per gene based on multiple probes the collapseRow function implemented in the WGCNA R package [48, 50, 83] was used to summarize the probe intensities. The resulting microarray expression matrix contained 33,204 genes or probe sets (i.e. where no map** to genomic location was found).
To finally construct a genome wide rice co-expression network, the following approach was conducted: Initially, the pairwise similarities of all 33,204 genes or probe sets based on the expression profiles across the 29 tissues were quantified using PCC. Further, the approach developed by Langfelder and Horvath [50] is used to derive a weighted co-expression network. More specifically, a similarity matrix S was constructed in which the entry Sij corresponds to the absolute value of the pairwise PCC:
where xi and xj represent of the expression profiles for genes or probe sets i and j,respectively.
Furthermore, the similarity matrix S was transformed into a weighted adjacency matrix, denoted by A. Here, the entry Aij is obtained by raising the previously derived co-expression similarity Sij to a power, β, β > =1:
The power, β, used to transform the similarity matrix is selected such that to the resulting network (described by its adjacency matrix) best approximates a scale-free topology – a defining network property of biological networks [84, 85]. In the case of the rice genome wide co-expression network, the parameter β = 7 was chosen (Additional file 10).
Gene modules were defined as sets of nodes in the co-expression network, i.e. genes and probe sets, with a high topological overlap [50, 51]. The topological overlap measure (TOM) between the ith and jth node is defined as
where denotes the number of nodes to which both nodes i and j are connected by an edge, denotes the sum of edge weights, i.e. the connection strengths, between ith gene and the other genes. Further, 1-TOM denotes the TOM based dissimilarity measure (1-TOM) which was used for hierarchical clustering. Finally, gene modules are obtained by using dynamic tree cutting algorithm on the resulting dendrogram. This outlined procedure were carried out using the blockwiseModule method implemented in the WGCNA R package (parameters: maxBlockSize = 20000, power = 7, minModuleSize = 50, reassignThreshold = 0, mergeCutHeight = 0.20) [83].
Connectivity scores of rice genes
Highly connected nodes in a network, commonly termed hubs, are thought to play a central role in the case of biological networks. The connectivity of a node has been used as a defining property for finding biologically relevant genes in co-expression networks [54]. Here, the intra-modular connectivity (kWithin) is used as a measure of centralization of genes. It is defined as the degree of the node corresponding to a gene inside a given module of the genome wide rice co-expression network [54]. The parameter kTotal was defined as the whole network connectivity for genes, reported as the sum of its connection strengths with all other genes in the network. A module’s eigengene-based connectivity (kME) was defined as the correlation between a gene expression value and the module eigengene (the average module expression value for an individual), which can be derived using R function consensusKME in the WGCNA package [50, 55].
Plant material collection and cell wall composition determination
The 29 tissues, or organs, of Zhenshan 97, indica variety were harvested at 16:00–18:00 of the day according to Wang et al. [44]. All samples were dried at 50°C after inactivation at 105°C for 5 min. The dried tissues were ground through a 40 mesh screen and stored in a dry container until use.
Plant cell wall fractionation procedure and cell wall composition analysis were described by Peng et al. [56] with modification by Li et al. [58]. The crude cell wall material was suspended in 0.5% (w/v) ammonium oxalate and heated for 1 h in a boiling water bath (supernatant referred to as pectins). The remaining pellet was first re-suspended in 4 M KOH containing 1.0 mg/mL sodium borohydride for 1 h at 25°C., and then the combined supernatant was neutralized, dialyzed and lyophilized (referred to as hemicelluloses). The non-KOH-extractable residue defined as crude cellulose, was further extracted with acetic:nitric acids:water (8:1:2) for 1 h at 100°C, and the remaining pellet was defined as cellulose. Cellulose was analyzed by anthrone/H2SO4 method. Monosaccharides (xylose, arabinose, galactose) of hemicelluloses were determined by GC-MS [58].
Three monolignols were detected by HPLC [57]. All the samples were extracted with benzene:ethanol (2:1, v/v) in a Soxhlet for 4 h, the remaining pellet was then collected as cell wall residue (CWR). The procedure of nitrobenzene oxidation of lignin was carried out as follows: First, 0.05 g CWR was added with 5 mL 2 M NaOH and 0.5 mL nitrobenzene, and a stir bar was put into a 25 mL Teflon gasket in a stainless steel bomb, and the bomb was sealed tightly and heated at 170°C (oil bath) for 3.5 h and stirred at 20 rpm. Then, the bomb was cooled with cold water, the chromatographic internal standard (ethyl vanillin) was added to the oxidation mixture. To remove nitrobenzene and its reduction byproducts, the alkaline oxidation mixture was washed 3 times with 30 mL CH2CI2/ethyl acetate mixture (1/1, v/v).The alkaline solution was acidified to pH 3.0-4.0 with 6 M HCl, and then extracted with CH2CI2/ethyl acetate (3 × 30 mL) to obtain the lignin oxidation products which were in the organic phase. The organic extracts were evaporated to dryness under reduced pressure 40°C. Finally, the oxidation products were dissolved in 10 mL chromatographic pure methanol. All experiments were carried out in triplicate. Standard chemicals: p-Hydroxybenzaldehyde(H), vanillin(G) and syringaldehyde (S) were purchased from Sinopharm Chemical Reagent Co., Ltd.
Identification of the cell wall-related modules through functional enrichment
GO terms of probes and genes were derived from agriGO [86]. To elucidate key biological processes, rather than conserved particular molecular functions, the GO sub-ontology 'biological process’ (GO-BP) was used for the gene-set enrichment analysis [87]. The enrichment analysis of particular GO-BP terms was performed using a weighted method in combination with Fisher’s exact test which is provided by topGO package [52]. KEGG ontology (KO) from the KEGG database (http://www.genome.jp/kegg/) [53] was additionally obtained and RAP IDs were converted to TIGR IDs using the RAP-DB ID converter tool (http://rapdb.dna.affrc.go.jp/tools/converter) [88]. KO enrichment was calculated by using hyper geometric test [89].
Analysis of the cell wall-related modules through physiologic traits
For each gene module, the module eigengenes, i.e. the first principle component of the expression profiles of all the modules members, was derived as a representative expression profile for each module. Module eigengenes were calculated through the WGCNA R package [50, 83]. Subsequently, the association of module eigengenes and the measured physiological traits was determined as follows: for each module, the eigengene (ME) was tested for significant associations with the external traits. In case such an association is present, subsequently, a correlation analysis was performed between all of the modules genes and the cell wall components individually to study finer substructure of particular gene/external trait relationships. In addition to the degree of correlation to the trait, the genes intra-modular connectivity is used to rank putative gene candidates.
Canonical correlations of cell wall traits and modules eigengenes
The set of cell wall features is represented by the matrix X in which rows correspond to the 29 tissues and columns correspond to the 7 cell wall measurements. Likewise, matrix Y of denotes the set of eigengenes whereas rows also correspond to the 29 tissues and columns correspond to the obtained 56 eigengenes. In CCA, and denote the two basis vectors, such that the correlation between the projections of the variables – columns in X and Y – onto these basis vectors given by and are mutually maximized: .
These derived linear projections U1 and V1 are called the first canonical variates. To investigate association between individual variables, i.e. eigengenes and cell wall features the similarity between variables in X and Y is quantified based on the Pearson correlations of their initial representation and the determined canonical variates. This form of correlations is known as canonical structure correlations [90] and can be further visualized by means of a relevance network [91]. Both, the CCA analysis as well the network are derived using the mixOmics package (http://www.math.univ-toulouse.fr/~biostat/mixOmics/) [92]. As a threshold for deriving edges between eigengenes and cell wall features in the relevance network, τ CCA ≥ 0.5 for the absolute values of association between variables was chosen further ensuring that all 7 cell wall parameters are not isolated in this network.
Availability of supporting data
All data sets supporting the results of this article are included within the article and also provided in the repository hosted by LabArchives, LLC (http://www.labarchives.com/) with DOI: http://dx.doi.org/10.6070/H4NV9G6V.
References
Lee I, Ambaru B, Thakkar P, Marcotte EM, Rhee SY: Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol. 2010, 28: 149-156.
Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P: Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004, 14: 1085-1094.
Persson S, Wei H, Milne J, Page GP, Somerville CR: Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc Natl Acad Sci U S A. 2005, 102: 8633-8638.
Stuart JM, Segal E, Koller D, Kim SK: A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003, 302: 249-255.
Ruprecht C, Mutwil M, Saxe F, Eder M, Nikoloski Z, Persson S: Large-scale co-expression approach to dissect secondary cell wall formation across plant species. Front Plant Sci. 2011, 2: 23-
Atias O, Chor B, Chamovitz D: Large-scale analysis of Arabidopsis transcription reveals a basal co-regulation network. BMC Syst Biol. 2009, 3: 86-
Mao L, Van Hemert JL, Dash S, Dickerson JA: Arabidopsis gene co-expression network and its functional modules. BMC Bioinformatics. 2009, 10: 346-
Mentzen WI, Peng J, Ransom N, Nikolau BJ, Wurtele ES: Articulation of three core metabolic processes in Arabidopsis: fatty acid biosynthesis leucine catabolism and starch metabolism. BMC Plant Biol. 2008, 8: 76-
Mutwil M, Usadel B, Schütte M, Loraine A, Ebenhöh O, Persson S: Assembly of an interactive correlation network for the Arabidopsis genome using a novel heuristic clustering algorithm. Plant Physiol. 2010, 152: 29-43.
Wang Y, Hu Z, Yang Y, Chen X, Chen G: Function annotation of an SBP-box gene in Arabidopsis based on analysis of co-expression networks and promoters. Int J Mol Sci. 2009, 10: 116-132.
Wei H, Persson S, Mehta T, Srinivasasainagendra V, Chen L, Page GP, Somerville CR, Loraine A: Transcriptional coordination of the metabolic network in Arabidopsis. Plant Physiol. 2006, 142: 762-774.
Faccioli P, Provero P, Herrmann C, Stanca A, Morcia C, Terzi V: From single genes to co-expression networks: extracting knowledge from barley functional genomics. Plant Mol Biol. 2005, 58: 739-750.
Ficklin SP, Luo F, Feltus FA: The association of multiple interacting genes with specific phenotypes in rice using gene coexpression networks. Plant Physiol. 2010, 154: 13-24.
Lee TH, Kim YK, Pham TTM, Song SI, Kim JK, Kang KY, An G, Jung KH, Galbraith DW, Kim M: RiceArrayNet: a database for correlating gene expression from transcriptome profiling and its application to the analysis of coexpressed genes in rice. Plant Physiol. 2009, 151: 16-33.
Ogata Y, Suzuki H, Sakurai N, Shibata D: CoP: a database for characterizing co-expressed gene modules with biological information in plants. Bioinformatics. 2010, 26: 1267-1268.
Edwards KD, Bombarely A, Story GW, Allen F, Mueller LA, Coates SA, Jones L: TobEA: an atlas of tobacco gene expression from seed to senescence. BMC Genomics. 2010, 11: 142-
Ficklin SP, Feltus FA: Gene coexpression network alignment and conservation of gene modules between two grass species: maize and rice. Plant Physiol. 2011, 156: 1244-1256.
Manfield IW, Jen CH, Pinney JW, Michalopoulos I, Bradford JR, Gilmartin PM, Westhead DR: Arabidopsis Co-expression Tool (ACT): web server tools for microarray-based gene expression analysis. Nucleic Acids Res. 2006, 34: W504-W509.
Obayashi T, Hayashi S, Saeki M, Ohta H, Kinoshita K: ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res. 2009, 37: D987-D991.
Vandepoele K, Quimbaya M, Casneuf T, De Veylder L, Van de Peer Y: Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol. 2009, 150: 535-546.
Mutwil M, Klie S, Tohge T, Giorgi FM, Wilkins O, Campbell MM, Fernie AR, Usadel B, Nikoloski Z, Persson S: PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell. 2011, 23: 895-910.
Higashi Y, Saito K: Network analysis for gene discovery inplant specialized metabolism. Plant Cell Environ. 2013, 36: 1587-1606.
Somerville CR, Bauer S, Brininstool G, Facette M, Hamann T, Milne J, Osborne E, Paredez A, Persson S, Raab T: Toward a systems approach to understanding plant cell walls. Science. 2004, 306: 2206-2211.
Zhang W, Yi Z, Huang J, Li F, Hao B, Li M, Hong S, Lv Y, Sun W, Arthur R, Hu F, Peng J, Peng L: Three lignocellulose features that distinctively affect biomass enzymatic digestibility under NaOH and H2SO4 pretreatments in Miscanthus. Bioresour Technol. 2013, 130: 30-37.
Smook GA: Handbook for Pulp and Paper Technologists. 1992, Vancouver: Angus Wilde Publications, 163-183.
Scheller HV, Ulvskov P: Hemicelluloses. Annu Rev Plant Biol. 2010, 61: 263-289.
Zhao Q, Dixon RA: Transcriptional networks for lignin biosynthesis: more complex than we thought?. Trends Plant Sci. 2011, 16: 227-233.
Sun H, Li Y, Feng S, Zou W, Guo K, Fan C, Si S, Peng L: Analysis of five rice 4-coumarate: coenzyme A ligase enzyme activity and stress response for potential roles in lignin and flavonoid biosynthesis in rice. Biochem Biophys Res Commun. 2012, 430: 1151-1156.
Carpita N, Tierney M, Campbell M: Molecular biology of the plant cell wall: searching for the genes that define structure architecture and dynamics. Plant Mol Biol. 2001, 47: 1-5.
Torney F, Moeller L, Scarpa A, Wang K: Genetic engineering approaches to improve bioethanol production from maize. Curr Opin Biotechnol. 2007, 18: 193-199.
**e G, Peng L: Genetic engineering of energy crops: a strategy for biofuel production in China. J Integr Plant Biol. 2011, 53: 143-150.
Turner SR, Somerville CR: Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant Cell. 1997, 9: 689-701.
Arioli T, Peng L, Betzner AS, Burn J, Wittke W, Herth W, Camilleri C, Höfte H, Plazinski J, Birch R: Molecular analysis of cellulose biosynthesis in Arabidopsis. Science. 1998, 279: 717-720.
Taylor NG, Scheible WR, Cutler S, Somerville CR, Turner SR: The irregular xylem3 locus of Arabidopsis encodes a cellulose synthase required for secondary cell wall synthesis. Plant Cell. 1999, 11: 769-779.
Taylor NG, Laurie S, Turner SR: Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis. Plant Cell. 2000, 12: 2529-2539.
Burk DH, Liu B, Zhong R, Morrison WH, Ye Z: A Katanin-like Protein Regulates Normal Cell Wall Biosynthesis and Cell Elongation. Plant Cell. 2001, 13: 807-827.
Peng L, Kawagoe Y, Hogan P, Delmer D: Sitosterol-beta-glucoside as primer for cellulose synthesis in plants. Science. 2002, 295: 147-150.
Zhong R, Burk DH, Morrison WH, Ye Z: A kinesin-like protein is essential for oriented deposition of cellulose microfibrils and cell wall strength. Plant Cell. 2002, 14: 3101-3117.
Brown DM, Zeef LAH, Ellis J, Goodacre R, Turner SR: Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell. 2005, 17: 2281-2295.
Brown DM, Goubet F, Wong VW, Goodacre R, Stephens E, Dupree P, Turner SR: Comparison of five xylan synthesis mutants reveals new insight into the mechanisms of xylan synthesis. Plant J. 2007, 52: 1154-1168.
Li A, **a T, Xu W, Chen T, Li X, Fan J, Wang R, Feng S, Wang Y, Wang B, Peng L: An integrative analysis of four CESA isoforms specific for fiber cellulose production between Gossypium hirsutum and Gossypium barbadense. Planta. 2013, 237: 1585-1597.
Wang L, Guo K, Li Y, Tu Y, Hu H, Wang B, Cui X, Peng L: Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010, 10: 282-
**e G, Yang B, Xu Z, Li F, Guo K, Zhang M, Wang L, Zou W, Wang Y, Peng L: Global identification of multiple OsGH9 family members and their involvement in cellulose crystallinity modification in rice. PLoS One. 2013, 8: e50171-
Wang L, **e W, Chen Y, Tang W, Yang J, Ye R, Liu L, Lin Y, Xu C, **ao J, Zhang Q: A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010, 61: 752-766.
Li Y, Qian Q, Zhou Y, Yan M, Sun L, Zhang M, Fu Z, Wang Y, Han B, Pang X, Chen M, Li J: Brittle culm1 which encodes a cobra-like protein affects the mechanical properties of rice plants. Plant Cell. 2003, 15: 2020-2031.
Zhou Y, Li S, Qian Q, Zeng D, Zhang M, Guo L, Liu X, Zhang B, Deng L, Liu X, Luo G, Wang X, Li J: BC10 a DUF266-containing and Golgi-located type II membrane protein is required for cell-wall biosynthesis in rice (Oryza sativa L). Plant J. 2009, 57: 446-462.
Zhang B, Zhou Y: Rice brittleness mutants: a way to open the 'black box’of monocot cell wall biosynthesis. J Integr Plant Biol. 2011, 53: 136-142.
Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, Horvath S: Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011, 12: 322-
R Development Core Team: R: A Language and Environment for Statistical Computing. version 2.13.1. 2012, Vienna Austria: R Foundation for Statistical Computing
Langfelder P, Horvath S: WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008, 9: 559-
Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL: Hierarchical organization of modularity in metabolic networks. Science. 2002, 297: 1551-1555.
Alexa A, Rahnenfuhrer J, Lengauer T: Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006, 22: 1600-1607.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38: D355-D360.
Dong J, Horvath S: Understanding network concepts in modules. BMC Syst Biol. 2007, 1: 24-
Langfelder P, Mischel PS, Horvath S: When is hub gene selection better than standard meta-analysis?. PLoS One. 2013, 8: e61505-
Peng L, Hocart CH, Redmond JW, Williamson RE: Fractionation of carbohydrates in Arabidopsis root cell walls shows that three radial swelling loci are specifically involved in cellulose production. Planta. 2000, 211: 406-414.
Xu N, Zhang W, Ren S, Liu F, Zhao C, Liao H, Xu Z, Huang J, Li Q, Tu Y, Yu B, Wang Y, Jiang J, Qin J, Peng L: Hemicelluloses negatively affect lignocellulose crystallinity for high biomass digestibility under NaOH and H2SO4 pretreatments in Miscanthus. Biotechnol Biofuels. 2012, 5: 58-
Li F, Ren S, Zhang W, Xu Z, **e G, Chen Y, Tu Y, Li Q, Zhou S, Li Y, Tu F, Liu L, Wang Y, Jiang J, Qin J, Li S, Li Q, **g H, Zhou F, Gutterson N, Peng L: Arabinose substitution degree in xylan positively affects lignocellulose enzymatic digestibility after various NaOH/H2SO4 pretreatments in Miscanthus. Bioresour Technol. 2013, 130: 629-637.
Usadel B, Obayashi T, Mutwil M, Giorgi FM, Bassel GW, Tanimoto M, Chow A, Steinhauser D, Persson S, Provart NJ: Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ. 2009, 32: 1633-1651.
Yan C, Yan S, Zeng X, Zhang Z, Gu M: Fine map** and isolation of bc7(t), allelic to OsCesA4. J Genet Genom. 2007, 34: 1019-1027.
Zhang B, Deng L, Qian Q, **ong G, Zeng D, Li R, Guo L, Li J, Zhou Y: A missense mutation in the transmembrane domain of CESA4 affects protein abundance in the plasma membrane and results in abnormal cell wall biosynthesis in rice. Plant Mol Biol. 2009, 71: 509-524.
Hussey SG, Mizrachi E, Spokevicius AV, Bossinger G, Berger DK, Myburg AA: SND2, a NAC transcription factor gene, regulates genes involved in secondary cell wall development in Arabidopsis fibres and increases fibre cell area in Eucalyptus. BMC Plant Biol. 2011, 11: 173-
Zhou J, Lee C, Zhong R, Ye Z: MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. Plant Cell. 2009, 21: 248-266.
Zhong R, Lee C, Ye Z: Global analysis of direct targets of secondary wall NAC master switches in Arabidopsis. Mol Plant. 2010, 3: 1087-1103.
Hirano K, Kondo M, Aya K, Miyao A, Sato Y, Antonio BA, Namiki N, Nagamura Y, Matsuoka M: Identification of transcription factors involved in rice secondary cell wall formation. Plant Cell Physiol. 2013, 54: 1791-1802.
Yoshida K, Sakamoto S, Kawai T, Kobayashi Y, Sato K, Ichinose Y, Yaoi K, Akiyoshi-Endo M, Sato H, Takamizo T, Ohme-Takagi M, Mitsuda N: Engineering the Oryza sativa cell wall with rice NAC transcription factors regulating secondary wall formation. Front Plant Sci. 2013, 4: 383-
Lee C, Zhong R, Ye Z: Arabidopsis family GT43 members are xylan xylosyltransferases required for the elongation of the xylan backbone. Plant Cell Physiol. 2012, 53: 135-143.
Brown DM, Wightman R, Zhang Z, Gomez LD, Atanassov I, Bukowski JP, Tryfona T, McQueen-Mason SJ, Dupree P, Turner SR: Arabidopsis genes IRREGULAR XYLEM (IRX15) and IRX15L encode DUF579-containing proteins that are essential for normal xylan deposition in the secondary cell wall. Plant J. 2011, 66: 401-413.
Singh A, Singh U, Mittal D, Grover A: Transcript expression and regulatory characteristics of a rice glycosyltransferase OsGT61-1 gene. Plant Sci. 2010, 179: 114-122.
Brown DM, Zhang Z, Stephens E, Dupree P, Turner SR: Characterization of IRX10 and IRX10-like reveals an essential role in glucuronoxylan biosynthesis in Arabidopsis. Plant J. 2008, 57: 732-746.
Lee C, Teng Q, Huang W, Zhong R, Ye Z: The Arabidopsis family GT43 glycosyltransferases form two functionally nonredundant groups essential for the elongation of glucuronoxylan backbone. Plant Physiol. 2010, 153: 526-541.
Jones L, Ennos AR, Turner SR: Cloning and characterization of irregular xylem4 (irx4): a severely lignin-deficient mutant of Arabidopsis. Plant J. 2001, 26: 205-216.
Romano JM, Dubos C, Prouse MB, Wilkins O, Hong H, Poole M, Kang KY, Li E, Douglas CJ, Western TL, Mansfield SD, Campbell MM: AtMYB61 an R2R3-MYB transcription factor functions as a pleiotropic regulator via a small gene network. New Phytol. 2012, 195: 774-786.
Yamaguchi M, Demura T: Transcriptional regulation of secondary wall formation controlled by NAC domain proteins. Plant Biotechnol. 2010, 27: 237-242.
Yang SD, Seo PJ, Yoon HK, Park CM: The Arabidopsis NAC transcription factor VNI2 integrates abscisic acid signals into leaf senescence via the COR/RD genes. Plant Cell. 2011, 23: 2155-2168.
Zhong R, Lee C, Zhou J, McCarthy RL, Ye Z: A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008, 20: 2763-2782.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M: MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37: 914-939.
Yang C, Xu Z, Song J, Conner K, Barrena GV, Wilson ZA: Arabidopsis MYB26/MALE STERILE35 regulates secondary thickening in the endothecium and is essential for anther dehiscence. Plant Cell. 2007, 19: 534-548.
Pimrote K, Tian Y, Lu X: Transcriptional regulatory network controlling secondary cell wall biosynthesis and biomass production in vascular plants. Afr J Biotechnol. 2012, 11: 13928-13937.
Hotelling H: Relation between two sets of variates. Biometrika. 1936, 28: 321-377.
Kent WJ: BLAT–the BLAST-like alignment tool. Genome Res. 2002, 12: 656-664.
International Rice Genome Sequencing Project: The map-based sequence of the rice genome. Nature. 2005, 436: 793-800.
Langfelder P, Horvath S: Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007, 1: 54-
Barabasi AL, Albert R: Emergence of scaling in random networks. Science. 1999, 286: 509-512.
Zhang B, Horvath S: A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005, 4: 17-
Du Z, Zhou X, Ling Y, Zhang Z, Su Z: agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38: W64-W70.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005, 102: 15545-15550.
Tanaka T, Antonio BA, Kikuchi S, Matsumoto T, Nagamura Y, Numa H, Sakai H, Wu J, Itoh T, Sasaki T: The rice annotation project database (RAP-DB): 2008 update. Nucleic Acids Res. 2008, 36: D1028-
Rivals I, Personnaz L, Taing L, Potier MC: Enrichment or depletion of a GO category within a class of genes: which test?. Bioinformatics. 2007, 23: 401-407.
ter Braak CJF: Interpreting canonical correlation-analysis through biplots of structure correlations and weights. Psychometrika. 1990, 55: 519-531.
González I, Lê Cao KA, Davis MD, Déjean S: Insightful graphical outputs to explore relationships between two 'omics’ data sets. BioData Mining. 2013, 5: 19-
Lê Cao KA, González I, Déjean S: integrOmics: an R package to unravel relationships between two omics data sets. Bioinformatics. 2009, 25: 2855-2856.
Acknowledgements
This study was supported in part by grants from the 111 Project (B08032), the projects (31171524 & 31100140) of National Natural Science foundation of China, the China Postdoctoral Science Foundation (20090460964), the 973 Specific Pre-project (2010CB134401), the National Transgenic Project (2009ZX08009-119B) and Fundamental Research Funds for the Central Universities (2011PY047). S.P. and S.K. thank the Max-Planck Gesellschaft for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’contributions
KG performed all data collection and major analyses. YF, MZ, JZ and FT participated in chemical tests. WZ, SP. WZ, GX, LW, SK and YT participated in data interpretation. LP supervised the project and finalized the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2013_6285_MOESM1_ESM.ppt
Additional file 1: The distribution of probes mapped to genes. Columns in red enclosure indicate the different probes mapped to the same genes. Numbers of mapped probes are transformed as log. (PPT 110 KB)
12864_2013_6285_MOESM2_ESM.xls
Additional file 2: Gene modules with rice locus identifiers, kME value of interested modules, kTotal, kWithin and kOut for each gene or probe set.(XLS 8 MB)
12864_2013_6285_MOESM3_ESM.ppt
Additional file 3: Module eigenvector clustering and number of genes (probes) in each module. A. Distribution of genes (probes) in each module, Red line indicated number of 500 genes. B. The co-expression network with 56 modules, and the eigenvectors of each module, calculated and clustered using the WGCNA software. (PPT 128 KB)
12864_2013_6285_MOESM6_ESM.xls
Additional file 6: Genes/Probes and their orthologs involved in Module 44, 24, 40, 42, 46 with all k-values (kTotal,kWithin kOut and kME except 6B) and predict function were listed. Ara:Arabinose related function; Gal:Galatose synthesis;Multi:Mutile roles;NA: None mapped;NF:None ortholog genes. (XLS 226 KB)
12864_2013_6285_MOESM10_ESM.ppt
Additional file 10: Analysis of network topology through different soft-thresholding powers. Left panel displays the scale-free fit index as a function of the soft-thresholding power. Right panel shows the mean connectivity (degree) as a function of the soft-thresholding power. (PPT 62 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Guo, K., Zou, W., Feng, Y. et al. An integrated genomic and metabolomic framework for cell wall biology in rice. BMC Genomics 15, 596 (2014). https://doi.org/10.1186/1471-2164-15-596
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-596