
Abstract
Background
Tomato (Solanum lycopersicum) establishes a beneficial symbiosis with arbuscular mycorrhizal (AM) fungi. The formation of the mycorrhizal association in the roots leads to plant-wide modulation of gene expression. To understand the systemic effect of the fungal symbiosis on the tomato fruit, we used RNA-Seq to perform global transcriptome profiling on Moneymaker tomato fruits at the turning ripening stage.
Results
Fruits were collected at 55 days after flowering, from plants colonized with Funneliformis mosseae and from control plants, which were fertilized to avoid responses related to nutrient deficiency. Transcriptome analysis identified 712 genes that are differentially expressed in fruits from mycorrhizal and control plants. Gene Ontology (GO) enrichment analysis of these genes showed 81 overrepresented functional GO classes. Up-regulated GO classes include photosynthesis, stress response, transport, amino acid synthesis and carbohydrate metabolism functions, suggesting a general impact of fungal symbiosis on primary metabolisms and, particularly, on mineral nutrition. Down-regulated GO classes include cell wall, metabolism and ethylene response pathways. Quantitative RT-PCR validated the RNA-Seq results for 12 genes out of 14 when tested at three fruit ripening stages, mature green, breaker and turning. Quantification of fruit nutraceutical and mineral contents produced values consistent with the expression changes observed by RNA-Seq analysis.
Conclusions
This RNA-Seq profiling produced a novel data set that explores the intersection of mycorrhization and fruit development. We found that the fruits of mycorrhizal plants show two transcriptomic “signatures”: genes characteristic of a climacteric fleshy fruit, and genes characteristic of mycorrhizal status, like phosphate and sulphate transporters. Moreover, mycorrhizal plants under low nutrient conditions produce fruits with a nutrient content similar to those from non-mycorrhizal plants under high nutrient conditions, indicating that AM fungi can help replace exogenous fertilizer for fruit crops.
Similar content being viewed by others


Background
Tomato (Solanum lycopersicum), like other solanaceous species, establishes a beneficial root symbiosis with arbuscular mycorrhizal (AM) fungi of the Glomeromycota [1]. The plant roots accommodate the AM fungi, which improve nutrient uptake of their plant hosts, and, in turn, receive fixed carbon from the plant [2], with a reciprocal reward for the best partner [3]. Furthermore, research on tomato mycorrhizal roots has identified plant genes as functional markers for AM symbiosis and shown that they are expressed in different cell types [4, 5]. In addition to the expected effects on roots, mycorrhization also affects gene expression in tomato shoots and leaves, modulating genes involved in diverse metabolic processes such as defence, transport and hormonal metabolism [6, 7]. A pilot microarray experiment also demonstrated a systemic effect on the fruit transcriptome [8].
Tomato represents an excellent model plant for understanding the long-distance effects of AM fungi on organs other than roots, as it exhibits substantial increases in growth and fruit yield in response to mycorrhization [9–11]. In addition, extracts of tomato fruit produced from mycorrhizal plants showed an anti-oestrogenic activity likely attributable to their higher contents of lycopene [12].
Analysis of the tomato (Solanum lycopersicum) genome [13] has showed that many tomato genes are specifically related to the evolution of fleshy fruits, and that intensive breeding has selected some agriculturally relevant traits. Recent work has characterized the molecular mechanisms that govern the transition of tomato fruit from a green to a fully red ripe fruit, via the intermediate stages termed breaker, turning and pink. Tomato ripening involves increased levels of aromatic compounds and carotenoid pigments, softening of the cell wall and decreased photosynthetic activity. Ethylene, the key hormone regulating ripening in climacteric fruit, controls most of these changes through a complex mechanism [14, 15]. Accumulating data have helped to define the roles played by specific genes [16] in specific fruit cell types [17], and also have extended the analysis to tomato epigenetic and proteomic dynamics [18].
To better explore the intersection between mycorrhization and fruit development in tomato, we used RNA-Seq to profile transcription changes [19] in fruits originating from mycorrhizal plants, a condition which has so far not been analysed in a crop plant of high agricultural interest. We found that the fruits of mycorrhizal plants show regulation of transcripts characteristic of climacteric fleshy fruits and a core set of genes typically modulated by symbiosis. Moreover, our quantitative analyses showed that fruits from mycorrhizal and non-mycorrhizal fertilized plants have comparable levels of carotenoids, phenols, and minerals (i.e. phosphorus, sulfur and potassium) indicating that the AM symbiosis benefits fruit quality. Thus, AM fungi can serve as surrogates for fertilization, providing similar benefits at the root and fruit levels.
Results
Plant sampling for RNA-Seq
To examine the transcriptome of tomato fruits from mycorrhizal plants, we collected fruits from plants colonized by the AM fungus Funneliformis mosseae (previously named Glomus mosseae) and from non-inoculated plants. AM fungi enhance nutrient uptake by their hosts [20]; to control for nutrition-dependent effects, we provided increased nutrients to the non-inoculated plants, to produce two groups of plants that have a similar phenology and nutritional status. Thus, we watered the mycorrhizal plants (MYC plants) with a low-phosphorus (P), nitrogen (N) and sulfur (S) solution, which allowed high mycorrhization levels and healthy plant growth [8, 21] but we watered the control, non-mycorrhizal plants (CONT plants) with higher concentrations of P, S and N. The MYC and CONT tomato plants grown under these different conditions showed similar growth (Figure 1) and similar P, S and K contents in their fruits (Additional file 1: Table S1), suggesting that the mycorrhizal fungus successfully compensated for the low fertilization treatment. We also measured some phenological features of the plant, including fruit yield, and found that the MYC and CONT fruits showed comparable values (Additional file 1: Table S2). The one exception was fruiting time, which was longer for mycorrhizal plants, leading to the production of more fruit. By contrast, plants that were not inoculated with F. mosseae but were fertilized with a similar, low-P and -N regimen to MYC plants were strongly affected by the nutrient limitations, showing impaired growth (Figure 1) and delayed fruit production, leading to the production of fewer fruit.

Development of tomato plants at 77 days after transplanting. Left: non-inoculated, low-fertilized plants; middle: non-inoculated highly-fertilized control plants (CONT); right: Mycorrhizal low-fertilized plants (MYC).
To assess the effect of mycorrhization on a key stage of ripening, we collected fruits at the turning stage, when the green color starts to change on 10 to 30 percent of the fruit surface, indicating the beginning of carotenoid biosynthesis [22]. Because the MYC and CONT plants have comparable vegetative periods, we harvested fruits from both conditions at 55 days after flowering. The MYC plants showed a colonization frequency (F) of 65% and a mycorrhizal intensity (M) of 36% in the root system (Additional file 2: Figure S1). We observed no traces of fungal colonization in the CONT roots. Two biological replicates for each condition were collected, following the current RNA-Seq standards (http://encodeproject.org/ENCODE/protocols/dataStandards/ENCODE_RNAseq_Standards_V1.0.pdf). The samples were then used for deep single-end sequencing (read length: 100 bp).
Analyses of RNA-Seq data: read number, transcriptome coverage and total expressed genes
To identify differentially expressed transcripts, we first filtered the sequences, estimated the transcriptome coverage, and identified expressed genes. The raw reads (100 bases, single-end) obtained from Illumina HIseq (FASTERIS Co. Geneva, Switzerland) were filtered (Illumina passed-filter call) and further checked for sequence contaminants with the fastQC application. Contaminant-free, filtered reads ranging from 11 to 16 million for each sample (Table 1) were mapped with Bowtie/TopHat to the tomato genome sequence SL2.40 (ITAG2.3) [13]. Raw read counts were obtained from alignment files by counting with HTSeq software. Based on the sum of transcript lengths, as reported in the current SL2.40 annotation (41,393,518 bp; which does not currently account for transcript isoforms), we reached an average transcriptome coverage of 23x for each replicate. An RPKM (Reads per Kilobase per Million reads) cutoff value of 0.1 was set to declare a locus expressed, resulting in 21,530 loci above the expression threshold. We also performed GO functional analysis to identify the biological processes that were most represented by the expressed genes. The GOSLIM terms for biological process assignment to the total expressed genes showed that cellular protein modification, carbohydrate metabolic, transmembrane transport, translation and lipid metabolic processes were the most-represented biological categories (Additional file 2: Figure S2).
Differentially expressed genes and overrepresented categories
To identify differentially expressed genes (DEGs), we used the R package DESeq to evaluate the significance of differences in expression and to control for false discovery rate (FDR). Pearson correlation coefficients among biological replicates were subjected to the same treatment and were all above 0.95. The FDR threshold was set to 0.1 and gene dispersion values were calculated by fitting a curve according to the DESeq “fit-only” mode. Using these criteria, we identified 712 DEGs (Additional file 3: Table S3), including 458 up-regulated genes and 254 down-regulated genes. We also used MA plot analysis to examine the magnitude distribution of the significantly regulated genes comparing the expression level to the log-transformed fold-change between MYC and CONT (Additional file 2: Figure S3). Among the DEGs, the 5 most highly-expressed genes (regardless of the growth condition) were a fruit-specific protein, Linoleate 9S-lipoxygenase B, the acid beta-fructofuranosidase and two heat shock proteins 90 and 70 (Table 2). These genes affect important fruit processes like ripening, stress responses, and lipid and carbohydrate biosynthesis.
GO annotation of differentially expressed genes
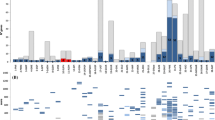
We used GO enrichment analysis to identify the major gene groups affected by mycorrhization. As in all RNA-Seq experiments, the variable length of transcripts may produce biases in the data [23]; therefore, we analyzed GO enrichment via the Goseq R package, which was specifically designed to limit bias in the data [24]. We found 81 GO terms that were over-represented (FDR ≤ 0.1) in response to mycorrhization (Figure 2). The biological processes corresponding to the lowest p-values, in decreasing order of importance, were “photosynthesis” (p = 2.66E-43), “photosynthesis light harvesting” (p = 4.48E-24), “protein-chromophore linkage” (p = 2.45E-17), “oxidation-reduction process” (p = 1.74E-13). Other relevant over-represented categories included: “response to biotic stimulus”, “transmembrane transport” and “carbohydrate metabolic process” (Figure 2).

GO enrichment in DEGs. The 81 enriched GO terms (y axis labels; FDR threshold ≤ 0.1) associated to DEG, as assessed compared with all expressed genes (RPKM > 0.1), are plotted along the reciprocal of Enrichment p value (x axis) as calculated with the Goseq R package. Panel A: Biological Process; panel B: Molecular Function; panel C: Cell Compartment.
Gene regulation in tomato fruit of mycorrhizal versusfertilized plants
Examination of GO terms suggested that a large part of the modulated tomato transcriptome is devoted to control of the main metabolic processes (i.e. photosynthesis and photorespiration) and to ripening (from cell-wall to secondary metabolites). For example, we identified 78 genes involved in oxidation-reduction processes, 54 in metabolic processes, 49 in photosynthesis, 43 with a catalytic activity, 30 involved in transport process, 24 transcription factors, 20 with a role in carbohydrate metabolism, 19 stress-responsive genes, 11 hormone-related genes and 51 unknown proteins corresponding to 10.95%, 7.58%, 6.88%, 6%, 4.17%, 3.37%, 2.8%, 2.66%, 1.52%, and 7.12% of the regulated gene core set, respectively. In the following sections, we focus on a subset of these genes.
Photosynthesis- and photorespiration-related processes
We identified a substantial number of genes involved in different steps of photosynthesis as up-regulated in MYC fruit (Table 3). Some of these up-regulated genes encode proteins of photosystem II and photosystem I, and others are involved in light-dependent reactions (e.g. cytochrome b6f, plastocyanins, ATP synthases and NADP reductases). Widespread up-regulation was also observed for genes related to the light-independent reactions or the Calvin cycle: two Ribulose bisphosphate carboxylases, one phosphofructokinase family protein, four fructose-1-6-bisphosphatases class 1 and three fructose-bisphosphate aldolases.
We also observed up-regulation of genes involved in chlorophyll metabolism. Six genes involved in chlorophyll biosynthesis and one gene involved in chlorophyll degradation (encoding Senescence-inducible chloroplast stay-green protein 2) were also found to be up-regulated. Genes involved in chlorophyll biosynthesis encoded proteins including: magnesium chelatase H subunit, magnesium-protoporphyrin IX methyltransferase, magnesium-protoporphyrin IX monomethyl ester, light dependent NADH:protochlorophyllide oxidoreductase 1, chlorophyllide a oxygenase and geranylgeranyl reductase. In this context, it was of particular interest to observe the up-regulation of the Golden 2-like transcription factor (SlGLK2) (log2 ratio of 1.5) (Additional file 3: Table S3). This gene belongs to the GARP family of MYB transcription factors, and determines chlorophyll accumulation and distribution in develo** fruit, thus influencing the entire photosynthetic process [25]. For chlorophyll degradation, we noted a slight up-regulation of the senescence-inducible chloroplast stay-green protein 1 (SlSGR1) in MYC fruit (log2 ratio of 1). Stay-green genes encode members of a family of chloroplast proteins, involved in the breakdown of chlorophyll–apoprotein complexes [26].
Genes involved in photorespiration (encoding serine hydroxymethyltransferase, hydroxypyruvate reductase, serine-glyoxylate aminotransferase, glycine decarboxylase and glycolate oxidase were also up-regulated (Additional file 3: Table S3) in MYC fruit, suggesting that the photorespiratory activity in the fruit was probably higher under the mycorrhizal conditions.
Taken as a whole, our transcript profiling data are consistent with numerous studies reporting the expression of genes involved in light and dark photosynthetic reactions, and photorespiration at turning and later ripening stages of the tomato fruit [13, 27–29]. Biochemical data showed a decrease in enzymatic activity of glycolate oxidase and hydroxypyruvate reductase from breaker to red ripe stages [27] while a time-course analysis of genome-wide gene expression [13] showed that the large majority of photosynthetic and respiratory genes identified in our experiment are down-regulated during the transition from green to red ripe fruit. Given that the nutritional status of the fruits from MYC and CONT plants was comparable, and all these genes were up-regulated in the MYC fruit, we hypothesize that mycorrhization may help extend the duration of the photosynthetic and photorespiratory activities in tomato fruit.
Carbohydrate and cell wall metabolism
Tomato fruit ripening involves accumulation of sugars and breakdown of cell wall carbohydrates, resulting in fruit softening. We identified twenty differentially regulated genes belonging to the carbohydrate metabolic process group, eight that are up-regulated in MYC fruit and twelve that are down-regulated (Table 4). Almost all genes involved in cell-wall carbohydrate modification were down-regulated (Figure 3) and many of them (glycoside hydrolase family 28 protein, beta-1-3-glucanase, beta-galactosidase, endo-1-4-beta-xylanase and polygalacturonase 2) are known to be involved in the degradation of cell-wall integrity, inducing fruit softening during ripening [30–33]. In the same category, the 5-xyloglucan endotransglucosylase/hydrolases (XTH) which encode enzymes with xyloglucan endotransglucosylase (XET) or endohydrolase (XEH) activities were also down-regulated. The role of this gene family during tomato ripening has been extensively investigated [34–36] and XTH genes show different behaviors during ripening. In particular, XTH1, XTH7, XTH12 and XTH6 had their highest expression in the mature green stage and then decreased in the later stages.

Clustering and heatmap of expression values for DEGs in the cell wall structure-related gene class. Light and dark blue indicate higher and lower expression values, respectively. Gene descriptions are as reported in Tomato Genome Consortium (2012). The heatmap was generated starting from DESeq normalized counts and further processed with custom scripts based on heatmap.2 function as available in the ‘gplots’ Bioconductor package and transformation function VST (DESEq package).
Our data also show the down-regulation of other genes encoding cell wall associated proteins such as pectinesterases and expansins (Table 4). Expansins are involved in expansion of plant cells in growing tissues but were also identified in non-growing tissues [37]. One of the expansins identified in our experiment is LeExp1 (Solyc06g051800.2.1) which was found to be specifically expressed in ripening fruit, with expression that was very weak at the green stage and increased with ripening [13]. In addition, LeExp1 seemed to be directly regulated by ethylene at the transcriptional level [37]. The second class of cell-wall modifying proteins identified was the pectinesterases. Pectinesterases demethylate pectins and make them more susceptible to polygalacturonase action [38], but they may also reinforce cell-to-cell adhesion [39]. The down-regulated pectinesterase gene identified in our experiment (PME 1.9) (Solyc07g064170.2.1) was one of the five most highly-expressed genes among the DEGs (Table 2).
All together, the results related to the regulation of genes encoding cell wall modifying enzymes show that, with few exceptions, the genes involved in cell wall hydrolysis are repressed in the MYC fruit, suggesting that the molecular mechanisms that lead to softening are delayed in the MYC fruit.
The MYC fruit also showed down-regulation of an acid beta-fructofuranosidase (Solyc03g083910.2.1; log2 ratio: -1.635) coupled with a higher up-regulation of a cell-wall invertase (Solyc10g083290.1.1; log2 ratio: 4.35) (Table 4). BLAST searches of these two sequences showed that they are, respectively, the tomato vacuolar invertase TIV1 and the cell-wall invertase (LIN6). These vacuolar and cell wall acid invertases, cleave the sucrose transported from source organs (leaves) into hexoses (glucose and fructose). Heterotrophic organs like fruit then use the hexoses as a direct source of carbon and energy [40]. The supply of sucrose to fruit and other sink tissues can limit their growth [41]. In carbohydrate supply to sink organs, sieve elements release sucrose into the apoplast where it undergoes an irreversible hydrolysis catalyzed by extracellular invertase and sucrose synthase [42]. The resulting hexoses are then transferred by hexose transporter(s) into sink cells. In tomato, extracellular invertases are encoded by LIN5, LIN6, LIN7 and LIN8, which have highly tissue-specific expression, regulated by internal and external signals [42]. TIV1 is expressed in red tomato fruit and LIN6 is highly expressed in seedling roots, flowers and tumors, but not in fruit [42]. Our RNA-Seq data show very low expression of LIN6 in CONT fruits (1.35 normalized read counts) but a much higher expression in MYC fruits (27.75 normalized read counts). Together, these data suggest that sucrose is actively mobilized in the MYC fruit, providing ready-to-use sugars for fruit development.
Transport processes
We identified 30 DEGs as involved in transport processes (Figure 4), and some of them are related to the DEGs involved in primary and carbohydrate metabolism. Among them, one of the most up-regulated genes in MYC fruit was a hexose transporter 2 (LeHT2) (Solyc09g075820.2.1; log2 ratio: 3.24). This gene shows the highest sequence similarity to a glucose/H + and low-affinity fructose/H + symporter that is expressed in tomato fruit storage parenchyma cells and strongly induced at fruit maturity [43, 44]. Hexose transporters transfer the hexoses hydrolyzed by cell wall invertases into sink cells. Therefore, they play an important role in sugar accumulation by fruit storage cells [45]. Indeed, a significant reduction (80–90%) in expression levels of three hexose transporter genes, LeHT1, LeHT2, LeHT3, by RNAi-mediated knockdown caused a 55% decrease in fruit hexose accumulation [44].

Clustering and heatmap of expression values for DEGs in the transmembrane transport-related gene class. Light and dark blue indicate higher and lower expression values, respectively. Descriptions as obtained from BLAST2GO annotation are shown. The heatmap was generated starting from DESeq normalized counts and further processed with custom scripts based on heatmap.2 function as available in the ‘gplots’ Bioconductor package and transformation function VST (DESEq package).
A tomato hexose transporter has been described as up-regulated after infection with Tomato yellow leaf curl virus and as co-regulated with a tomato lipocalin [46]. Lipocalins transport small hydrophobic molecules, and play important roles in various biological processes such as development and stress responses. Interestingly, a tomato lipocalin (Solyc07g005210.2.1) was activated in the MYC fruits, suggesting a network of interactions between transporters and responses to biotic stresses (see below).
We observed the up-regulation of one plasma membrane intrinsic protein and three tonoplast intrinsic protein aquaporins. Aquaporins function in water transport, and play pivotal roles in regulating water movements in fruit, which are crucial for fruit expansion and ripening [47]. Aquaporins are highly responsive to mycorrhization in legumes [48–52], although their exact role in the symbiosis is not clear [53] and tomato roots may also show a downregulation of aquaporins [6].
A urea transporter, characterized by a high up-regulation in the MYC fruit, showed 71% amino acid sequence similarity with an Arabidopsis thaliana sodium/urea cotransporter (DUR3). Interestingly, an allantoinase gene was found to be up-regulated in MYC fruit of the Micro-Tom variety, suggesting that the metabolism of urea precursors is also activated [8].
In accordance with the prominent function of transport in mycorrhizal roots, the membrane transporters functional category was found to be one of the most-regulated categories in roots colonized by AM fungi [50, 51, 54].
Our experiments identified two up-regulated genes involved in phosphate transport. The inorganic phosphate transporter (Solyc09g066410.1) has homology to both LePT1 and LePT3 phosphate transporters. Unlike LePT1, which is constitutively expressed [4], Solyc09g066410.1 seems to be induced by mycorrhization as it showed no basal expression in the fruit of non-inoculated plants. The second identified phosphate transporter (Solyc05g013510.2) shows the highest similarity with a chloroplastic inorganic phosphate transporter from S. tuberosum; this gene was highly expressed in young, rapidly growing tissues and may have a role in cell wall metabolism [55]. These data provide a good mechanistic basis for the observed fruit P content, which reaches comparable values in the two MYC and CONT conditions. This suggests that the mycorrhizal- dependent P uptake is as efficient as the P uptake from fertilization. Similarly, two sulphate-related transporters (High affinity sulphate transporter, see Additional file 3: Table S3) were more strongly expressed in the fruits from mycorrhizal plants, consistent with the comparable S content.
A solute carrier protein, probably acting as a nitrate transporter, was found to be down-regulated in our dataset. Nitrate transporters were found to be either up-or down-regulated in Medicago mycorrhizal roots, depending on the specific gene considered [48, 51, 56]. In tomato, colonization by F. mosseae specifically enhanced the expression of the nitrate transporter LeNRT2;3 without affecting the expression of other isoforms [57].
In conclusion, in addition to the expression of genes that function in fruit development, the presence of the AM fungus activates genes involved in nutrient and water transport activities, which are strictly related to the known functional markers of mycorrhizal roots.
Regulation of hormonal pathway genes
Tomato fruit development and ripening is stringently regulated by hormone dynamics [22]. In roots, the hormonal balance modulates mycorrhization and vice-versa [7, 58–60]. In addition, tomato genes involved in hormonal metabolism show a systemic response to the AM fungus [6]. We therefore expected hormone pathway genes to be differentially regulated in fruit; indeed, 11 genes were found to be involved in gibberellin, abscisic acid (ABA) and ethylene metabolism (Additional file 3: Table S3). A Gibberellin 2-oxidase 1 involved in the degradation of the active gibberellic acid (GA), and two Gibberellin regulated proteins, GAST1 (gibberellic acid stimulated transcript) and RSI-1 (Root system inducible-1), were found to be down-regulated in the fruit of MYC plants. Both GAST1 and RSI-1 belong to the GASA (GA-stimulated Arabidopsis) gene family, which includes many hormone-responsive genes involved in pathogen responses as well as in different aspects of plant development [61]. Two genes involved in ABA metabolism were found to be up-regulated in MYC fruit: the first one is zeaxanthin epoxidase, which catalyzes the conversion of zeaxanthin to violaxanthin [62], an ABA precursor [63]. The second gene is the ABA 8′-hydroxylase CYP707A2, which plays a role in ABA catabolism and is activated by the presence of ABA [64].
Six ethylene responsive factors were down-regulated in the MYC fruit (Additional file 3: Table S3): Ethylene response factor 1 (ERF1), a probable ethylene responsive transcription factor 2b, the C-repeat-binding factor 3 (CBF3), a probable ethylene-responsive transcription factor 1, the ethylene-responsive transcription factor 10 and the ethylene-responsive late embryogenesis-like protein. The ERFs are secondary ethylene activated factors, which increase plant tolerance to stress by modulating expression of downstream stress-responsive genes [65]. ERF1 is a key element in the response to different necrotrophic pathogens [66]. Interestingly, in Medicago the transcription factor ERF19 is down-regulated by the perception of an effector molecule secreted by an AM fungus [67]. However its most closely related homologue in tomato does not show differential expression during fruit ripening.
Overall, these data demonstrate the dynamic expression of genes involved in hormone metabolism: on the one hand, they mirror the transcriptional profile of fruit ripening markers (ethylene-responsive genes) and related pathways; on the other, some of them mirror the known role of hormone networks in AM establishment. Indeed, a positive correlation between ABA and mycorrhization in roots was reported [58] and ABA was found to be necessary for complete and fully functional arbuscule development and for successful colonization of the plant root [58]. By contrast, numerous studies have shown a reciprocal negative relationship between ethylene and mycorrhization [7, 59]. However, the role of hormone networks in AM establishment remains far from fully understood [7, 60].
Terpenoid metabolism
Terpenoids include phytohormones (abscisic acid, gibberellins, cytokinins, and brassinosteroids), photosynthetic pigments (phytol and carotenoids), and electron carriers (ubiquinone) [68]. However, most terpenoids are specialized secondary metabolites that function as defensive compounds accumulating upon pathogen infection (i.e. phytoalexins) [69] or attracting predators/parasitoids of the attacking insect [70]. In tomato, terpenoids are present in large quantities in the glandular trichomes on leaves, stems, young fruits, and flowers [71–74].
Three genes belonging to the terpene synthase (TPS) family, TPS31, TPS32 and TPS33, were induced in the tomato fruit by mycorrhization. Although only a few genes were affected, these three genes were among the ten most up-regulated by mycorrhization (Additional file 3: Table S3) and TPS31 showed no basal expression in CONT tomato fruits. TPS31, TPS32 and TPS33 belong to the sesquiterpenoid phytoalexin biosynthesis pathway (Additional file 2: Figure S4), leading to the production of the phytoalexin rishitin, well known for its antimicrobial properties [69]. Accumulation of phytoalexins has been suggested as a defense mechanism in mycorrhizal plant roots [75–77]. By contrast, genes involved in carotenoid synthesis did not show any change in their transcriptomic profiles (Additional file 2: Figure S5). These data were confirmed by a biochemical analysis of these metabolites, which showed no statistically significant differences between the two sets of fruits (Additional file 1: Table S4), with the exception of general reducing activity, which was higher in the MYC fruits.
Defense genes
Plants activate a wide range of mechanisms to defend themselves against external attacks [78]. Such responses are under the control of a finely tuned hormonal network, and can spread from the infection site to a systemic level. In our study, four out of the five up-regulated genes were pathogenesis related proteins (PRs) three PR10s, and one PR1. PRs specifically accumulate in plants in response to pathogen infection and act in development of systemic acquired resistance against further infections. PR regulation in response to mycorrhization appears to be complex; López-Ráez and colleagues [7] reported the induction of PR10 in tomato roots inoculated with Funneliformis mosseae and Rhizophagus intraradices. By contrast, some PRs were down-regulated in plant shoots following mycorrhization [6, 49] and induced only after a pathogen attack, as a result of the mycorrhizal priming effect [89]. During ripening, tomato fruits shift from partially photosynthetic to truly heterotrophic metabolism, as also mirrored by a decrease of many photosynthetic proteins together with those involved in chlorophyll biosynthesis [29]. However, notwithstanding the wealth of data, the role of fruit photosynthesis remains not fully understood [90]. Our transcriptomic analysis adds a novel piece of information to this emerging understanding, showing that mycorrhization might affect fruit photosynthesis and photorespiration. This transcriptional influence seems to be independent of the nutritional level, since mycorrhizal and control fertilized plants had similar mineral and nutrient contents in their fruits (Additional file 1: Table S1).
Here we found that tomato fruits of mycorrhizal plants sampled at the turning stage present an important up-regulation of photosynthetic genes, which are usually activated in immature green fruits [91]. Also, in MYC fruits photorespiration may be activated via the increased expression of a core set of genes (Additional file 2: Figure S2). AM fungi stimulate photosynthesis in leaves of different plants [92, 93]. Interestingly, strigolactones, which act as positive regulators of AM symbiosis, also up-regulate light-harvesting genes in the same tomato organs [94]. It has been proposed that mycorrhization causes an improvement of plant water status, increasing hydraulic conductance and affecting stomata and leaf expansion [95, 96]. Other investigations suggested that the AM fungi, which depend on their host for carbon, increase root sink strength and enhance leaf photosynthetic activity to compensate for the ‘cost’ imposed upon the plant C economy by the fungus [97, 98].
On the basis of our RNA-Seq profiling, we hypothesize that similar events also take place in the fruit, where the increased sink strength is confirmed by the activation of other metabolic pathways. The up-regulation of LIN6 in our MYC fruit is particularly intriguing as the expression of this gene has been reported to be high in roots, flowers and tumors and was almost absent in fruit [42]. Indeed, in our experiment, LIN6 was very weakly expressed in the CONT fruit.
It seems therefore that mycorrhization positively influences the expression of genes of the metabolic pathways related to carbon assimilation and respiration. In this context, the up-regulation of the SlGLK2 transcription factor [25] could provide novel and unexpected insights. SlGLK2 is encoded by the uniform ripening locus (u) and determines the intensity and pattern of chlorophyll distribution in the unripe fruit. The dominant U allele leads to fruit with dark green shoulders at the stem end adjacent to the pedicel, and u/u fruit are uniformly light green and were therefore favored by the breeders. However, in these fruits selection has led to suboptimal chloroplast development, and consequently, to decreased ripe fruit sugar and lycopene levels. It seems that in some way mycorrhization rescues the levels of SlGLK2 mRNA to higher levels, also probably controlling the downstream photosynthesis-related genes.
Could mycorrhization extend tomato shelf life?
The ripening of climacteric fruits such as tomato is characterized by a large burst of respiration known as the ‘climacteric rise’ at the onset of ripening. The climacteric rise is coupled with increased ethylene biosynthesis [39], which is thought to coordinate numerous biochemical and physiological processes necessary for normal ripening such as softening of the cell wall, accumulation of pigment and increases in flavor and aroma [14]. Cell wall softening, an important process influencing fruit quality and principally shelf life, involves a coordinated decline in the activity of cell-wall biosynthetic enzymes and an increase in the activity of cell wall hydrolytic enzymes, like polygalacturonase [99].
Availability of the high-quality, annotated tomato genome sequence [13] has allowed an extension of our understanding of ripening, identifying more than 40 genes involved in fruit cell wall architecture. The regulation of cell wall related genes strictly relates phenotypic features, including fruit firmness, and shelf life. Our transcriptomic data consistently demonstrate that mycorrhization may lead to a down-regulation of cell wall hydrolytic enzymes. Similar results were already reported in a previous, more limited study [8] of the Micro-Tom variety.
All these data, together with the up-regulation of the SlGLK2 transcription factor, the up-regulation of photosynthesis-related genes, the increased chlorophyll synthesis and the down-regulation of ethylene responsive genes, indicate that mycorrhization may lead to fruits with a potentially longer shelf-life through the activation of multiple pathways.
Conclusions
The extensive transcriptomic analysis we performed, comparing tomato fruits from mycorrhizal plants with fruits from fertilized plants, demonstrates that mycorrhization may have important systemic effects. By improving the nutritional status of the whole plant, mycorrhization leads to a mineral content in the fruits that is comparable to the mineral content of fruits from highly fertilized plants. By affecting the source-sink relationships of the whole plant, mycorrhization also has an impact on some of the features of fleshy fruits. This novel transcriptome survey thus shows biological intersections between roots and fruits, and will provide a platform for future, in-depth studies. The data we present here will allow study of the complex process of tomato fruit development under the usual cultivation conditions, like those occurring in greenhouses where the plants are fertilized, the soil is not sterile, and the complex tomato microbiome, also including AM fungi, affect plant performance.
Methods
Plant materials and growth conditions
Solanum lycopersicum cv. Moneymaker tomato plants were grown in the greenhouse from March through December under natural light, which provided a photoperiod ranging from 9 h to 15 h, temperature from 16°C to 30°C and relative humidity from 55% to 80%. The seedlings were grown in pots with sterile quartz sand (80%) and sterile pumice (20%) for non-inoculated plants or with a mixture of sand (55%), sterile pumice (20%) and Funneliformis mosseae (formerly Glomus mosseae) inoculum (25%) Gerd. & Trappe (BEG 12) purchased from Agrauxine (Bretagne, France, http://www.agrauxine.fr) for inoculated plants. Forty-five plants divided into three groups of fifteen plants according to their growth conditions were considered: plants grown with F. mosseae inoculum (MYC) or without inoculum were watered, once a week, with a modified Long-Ashton solution containing a low phosphorus and nitrogen concentration (3.2 μM P, 1 mM N, 1.75 mM S, 1 mM K) [100]. The fertilized control plants (CONT) were grown without inoculum and watered, once a week, with a commercial fertilizer solution (3.8 mM P, 12.9 mM N, 6.6 mM S, 3.8 mM K) (Asso di fiori, CIFO S.p.A). Phenological features were monitored during the experiments, as summarized in Additional file 1: Table S2. At the end of the experiment, roots from MYC and CONT plants were cut and fungal colonization was assessed according to the Trouvelot system [101] using MYCOCALC software.
Illumina sequencing
For the RNA-Seq experiment, fruits were harvested from the CONT and MYC plants when the fruits reached the turning stage. RNA was extracted using the ‘pine tree-method’ [102] with the addition of 1% PVPP to the extraction buffer. For each growth condition, we used two biological replicates, each containing the pooled RNA from three fruits. Ten micrograms of each RNA sample was sent to Fasteris Life Science Co. (Geneva, Switzerland) where the libraries were produced and sequenced using the Illumina Genome Analyzer (Solexa). The four libraries were indexed and single-end multiplexed sequencing was performed using 100 bp length reads.
Time course quantitative Real-Time PCR validation
Quantitative Real-Time PCR, was used to measure the expression of nine genes shown to be differentially regulated by RNA-Seq, as well as of five carotenoid related genes. Three fruit ripening stages were considered: mature green, breaker and turning. For each stage, three biological replicates were used. RNA extraction, primer design, quantitative real time PCR experiment and data analysis were carried out as described in Salvioli et al. [8]. The primer names and corresponding sequences are listed in Additional file 1: Table S5.
Bioinformatic methods
Map** of Illumina reads
Raw fastQ files were checked for contaminants and low-quality bases and contaminants were removed with the cutadapt software [103]. The spliced read mapper TopHat version 1.4.1 [104] was used to map reads to Tomato SL2.40 (ITAG2.3) obtained from the plant repository. A minimum and maximum intron length of 40 and 50,000 bp were used, respectively. Read counts were collected with HTSeq version 0.5.3 (http://www-huber.embl.de/users/anders/HTSeq) in the single end and ‘union’ mode using Solanum lycopersicum MSU SL2.40-16 GTF file as obtained from the ensembl plant repository.
DEG calling
The DESeq Bioconductor package version 1.10.1 [105] was used to call differentially expressed genes. DESeq implements a model based on negative binomial distribution; this model was developed with special attention to co** with biological variance and was run under R release 2.15.2. The cutoff for considering a gene expressed was set to 0.1 RPKM (Reads per Kilobase per Million). The False Discovery Rate (FDR) threshold was set to 0.1 and gene dispersion values were calculated by fitting a curve according to the DESeq “fit-only” mode.
GO enrichment analyses
GO enrichment analyses were conducted with the goseq bioconductor package version 1.10.0. As tomato databases are not yet covered by goseq built-in databases, transcripts lengths were retrieved with BiomaRt queries (Solanum lycopersicum SL2.40). An FDR cutoff of 0.1 was used for goseq GO enrichments. Gene ontology terms for CC, MF and BP were similarly retrieved with BiomaRT queries.
Other bioinformatic techniques
Pie charts for the Distribution of GOSLIM BP terms were generated via BLAST2GO software using SL2.40 annotations. MapMan figures were obtained by first running the mercator tool (http://mapman.gabipd.org/web/guest/mercator) with default parameters to assign MapMan bins to tomato transcripts. Log2 fold changes as obtained from DESeq output were used as MapMan input to represent expression changes. For clustering and heatmaps of expression values, DESeq-normalized data were prior transformed with the Variance-stabilizing transformation (VST) as described by Anders and Huber [105]. Unless otherwise stated, further graphical outputs were generated with custom R and Python scripts. Custom annotations/descriptions were used as specified in the text and figures and were obtained with BLAST2GO on tomato SL2.40 cDNAs run with the following annotation parameters: E-value hit filter 1.E-10, Annotation cutoff 55, Go weight 5, Hsp-Hit coverage cutoff 20. Further complementary descriptions (ITAG2.3_descriptions) were obtained from ftp://ftp.solgenomics.net/tomato_genome/annotation/ITAG2.3_release/.
Fruit mineral content measurements
About 5 g of frozen tomato fruit pericarp from MYC and CONT plants were dried at 70°C for 48 hours, and digested in 2 ml 6 M HNO3 at 90°C for one hour. The digestion product was diluted to 6 ml with milliQ water and filtered. The P, S and K contents were determined in the final solution using Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) performed using a Liberty 100 Varian apparatus equipped with a V-Groove nebulizer and a Czerny–Turner monochromator (Department of Mineralogical and Petrological Science, University of Turin). The same treatments were applied to the control solution, which contained no sample. Quality control was based on the use of internal control samples and certified samples (Astasol-Mix from Analytica Ltd., Prague, Czech Rep.). C and N content was measured on fruit powder with a CHNS-O analyzer (Thermo Electron Corporation).
Biochemical analyses for nutraceutical values
Fruit extraction
About 6 g of unfrozen fruit material were extracted on a magnetic stirrer for 15 min in 30 ml volume of a methanol, distilled water and formic acid (80:20:0.5) mixture. The extracts were double-filtered, first on blotting paper with a Büchner funnel connected to a vacuum pump and then with a 0.45 nm cellulose membrane. The total antioxidant activity and the phenolic content of the methanolic fruit extracts were immediately assayed. All spectrophotometric measurements were performed in glass cuvettes using a T80+ UV/VIS spectrometer (PG Instruments Ltd.).
Total antioxidant activity
The total antioxidant activity of the tomato fruit extracts was measured by testing the radical scavenging capacity (DPPH test) and the reducing power (FRAP assay), according to the methods described by Tenore et al. [106].
Total Phenolic content determination
The total phenolic content was analyzed spectrophotometrically using the modified Folin–Ciocalteu method [107, 108]. 200 μl of the fruit extract from MYC and CONT plants was mixed with 100 μl of Folin-Ciocalteu’s phenol reagent (Sigma) in a glass test tube. Successively, 5 ml of 7% sodium carbonate in distilled water was added to the tube and the mixture was kept in the dark at room temperature for 2 h. Absorbance was then measured at 760 nm and results were expressed as mg Gallic acid equivalents (GAE) per 100 g of tomato fresh weight. Gallic acid (Sigma) was used to calculate the standard curve at 0.025, 0.05, 0.1 and 0.15 mg/100 μl.
Lycopene and β-Carotene
Quantification of Lycopene and β-Carotene was performed spectrophotometrically according to the method described by Nagata and Yamashita [109]. One g of unfrozen fruit tissues from MYC and CONT plants was homogenized and extracted in 16 ml of an acetone/n-hexane 40:60 solution for 15 min on a magnetic stirrer. Extracts were then filtered using a 0.45 nm cellulose membrane and absorbance at 663, 645, 505 and 453 nm was measured. Contents of β-Carotene and Lycopene were calculated according to the following equations: Lycopene (mg/100 ml) = - 0.0458 A663 + 0.3jou72 A505 – 0.0806 A453; β-Carotene (mg/100 ml) = 0.216 A663 – 0.304 A505 + 0.452 A453. Results were finally expressed as mg/100 g fm.
Statistical analysis
For all the biochemical and nutraceutical measurements, five fruits were harvested for each MYC and CONT condition and originating from different plants. Values are expressed as mean ± standard deviation. Data were analyzed with a one-way ANOVA with Tukey post-hoc or the non parametric test Kruskal-Wallis accepting significant differences at p < 0.05. Significantly different values are highlighted in bold. All statistical analysis were performed with SYSTAT 10 statistical software.
Availability of supporting data
Supporting sequence data are available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under Accession number: E-MTAB-2276.
References
Schussler A, Schwarzott D, Walker C: A new fungal phylum, the Glomeromycota: phylogeny and evolution. Mycol Res. 2001, 105: 1413-1421. 10.1017/S0953756201005196.
Bonfante P, Genre A: Mechanisms underlying beneficial plant-fungus interactions in mycorrhizal symbiosis. Nat Commun. 2010, 1 (48): doi:10.1038/ncomms1046
Kiers ET, Duhamel M, Beesetty Y, Mensah JA, Franken O, Verbruggen E, Fellbaum CR, Kowalchuk GA, Hart MM, Bago A, Palmer TM, West SA, Vandenkoornhuyse P, Jansa J, Bücking H: Reciprocal rewards stabilize cooperation in the mycorrhizal symbiosis. Science. 2011, 333: 880-882. 10.1126/science.1208473.
Balestrini R, Gomez-Ariza J, Lanfranco L, Bonfante P: Laser microdissection reveals that transcripts for five plant and one fungal phosphate transporter genes are contemporaneously present in arbusculated cells. Mol Plant Microbe Interact. 2007, 20: 1055-1062. 10.1094/MPMI-20-9-1055.
Gomez-Ariza J, Balestrini R, Novero M, Bonfante P: Cell-specific gene expression of phosphate transporters in mycorrhizal tomato roots. Biol Fertil Soils. 2009, 45: 845-853. 10.1007/s00374-009-0399-2.
Fiorilli V, Catoni M, Miozzi L, Novero M, Accotto GP, Lanfranco L: Global and cell-type gene expression profiles in tomato plants colonized by an arbuscular mycorrhizal fungus. New Phytol. 2009, 184: 975-987. 10.1111/j.1469-8137.2009.03031.x.
Lopez-Raez JA, Verhage A, Fernandez I, Garcia JM, zcon-Aguilar C, Flors V, Pozo MJ: Hormonal and transcriptional profiles highlight common and differential host responses to arbuscular mycorrhizal fungi and the regulation of the oxylipin pathway. J Exp Bot. 2010, 61: 2589-2601. 10.1093/jxb/erq089.
Salvioli A, Zouari I, Chalot M, Bonfante P: The arbuscular mycorrhizal status has an impact on the transcriptome profile and amino acid composition of tomato fruit. BMC Plant Biol. 2012, 12: 4-10.1186/1471-2229-12-4. doi:10.1186/1471-2229-12-44
Bryla DR, Koide RT: Mycorrhizal response of two tomato genotypes relates to their ability to acquire and utilize phosphorus. Ann Bot. 1998, 82 (6): 849-857. 10.1006/anbo.1998.0768.
Poulton JL, Bryla D, Koide RT, Stephenson AG: Mycorrhizal infection and high soil phosphorus improve vegetative growth and the female and male functions in tomato. New Phytol. 2002, 154: 255-264. 10.1046/j.1469-8137.2002.00366.x.
Nzanza B, Marais D, Soundy P: Tomato (Solanum lycopersicum L.) seedling growth and development as influenced by Trichoderma harzianum and arbuscular mycorrhizal fungi. Afr J Microbiol Res. 2011, 5: 425-431.
Giovannetti M, Avio L, Barale R, Ceccarelli N, Cristofani R, Iezzi A, Mignolli F, Picciarelli P, Pinto B, Reali D, Sbrana C, Scarpato R: Nutraceutical value and safety of tomato fruits produced by mycorrhizal plants. Br J Nutr. 2012, 107: 242-251. 10.1017/S000711451100290X.
Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, Egholm M, Knight J, Bogden R, Li C, Shuang Y, Xu X, Pan S, Cheng S, Liu X, Ren Y, Wang J, Albiero A, Dal Pero F, Todesco S, Van Eck J, Buels RM, Bombarely A, Gosselin JR, Huang M, Leto JA, et al: The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012, 485: 635-641. 10.1038/nature11119.
Giovannoni J: Molecular biology of fruit maturation and ripening. Annu Rev Plant Physiol Plant Mol Biol. 2001, 52: 725-749. 10.1146/annurev.arplant.52.1.725.
Giovannoni JJ: Genetic regulation of fruit development and ripening. Plant Cell. 2004, 16: S170-S180. 10.1105/tpc.019158.
Luo Z, Zhang J, Li J, Yang C, Wang T, Ouyang B, Li H, Giovannoni J, Ye Z: A STAY-GREEN protein SlSGR1 regulates lycopene and beta-carotene accumulation by interacting directly with SlPSY1 during ripening processes in tomato. New Phytol. 2013, 198: 442-452. 10.1111/nph.12175.
Matas AJ, Yeats TH, Buda GJ, Zheng Y, Chatterjee S, Tohge T, Ponnala L, Adato A, Aharoni A, Stark R, Fernie AR, Fei Z, Giovannoni JJ, Rose JK: Tissue- and Cell-Type Specific Transcriptome Profiling of Expanding Tomato Fruit Provides Insights into Metabolic and Regulatory Specialization and Cuticle Formation. Plant Cell. 2011, 23: 3893-3910. 10.1105/tpc.111.091173.
Lopez-Casado G, Covey PA, Bedinger PA, Mueller LA, Thannhauser TW, Zhang S, Fei Z, Giovannoni JJ, Rose JK: Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics. 2012, 12: 761-774. 10.1002/pmic.201100164.
Van Verk MC, Hickman R, Pieterse CM, Van Wees SC: RNA-Seq: revelation of the messengers. Trends Plant Sci. 2013, 18: 175-179. 10.1016/j.tplants.2013.02.001.
Smith SERD: Mycorrhizal symbiosis. 2008, New York: Academic Press
Volpe V, Dell’Aglio E, Giovannetti M, Ruberti C, Costa A, Genre A, Guether M, Bonfante P: An AM-induced, MYB-family gene of Lotus japonicus (LjMAMI) affects root growth in an AM-independent manner. Plant J. 2013, 73: 442-455. 10.1111/tpj.12045.
Gillaspy G, Bendavid H, Gruissem W: Fruits - a developmental perspective. Plant Cell. 1993, 5: 1439-1451.
Oshlack A, Wakefield MJ: Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009, 4 (14): doi:10.1186/1745-6150-4-14
Young MD, Wakefield MJ, Smyth GK, Oshlack A: Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010, 11 (R14): doi:10.1186/gb-2010-11-2-r14
Powell AL, Nguyen CV, Hill T, Cheng KL, Figueroa-Balderas R, Aktas H, Ashrafi H, Pons C, Fernández-Muñoz R, Vicente A, Lopez-Baltazar J, Barry CS, Liu Y, Chetelat R, Granell A, Van Deynze A, Giovannoni JJ, Bennett AB: Uniform ripening encodes a golden 2-like transcription factor regulating tomato fruit chloroplast development. Science. 2012, 336: 1711-1715. 10.1126/science.1222218.
Hoertensteiner S: Stay-green regulates chlorophyll and chlorophyll-binding protein degradation during senescence. Trends Plant Sci. 2009, 14: 155-162. 10.1016/j.tplants.2009.01.002.
Martin BA, Gauger JA, Tolbert NE: Changes in activity of ribulose-1,5-bisphosphate carboxylase/oxygenase and three peroxisomal enzymes during tomato fruit development and ripening. Plant Physiol. 1979, 63: 486-489. 10.1104/pp.63.3.486.
Piechulla B, Glick RE, Bahl H, Melis A, Gruissem W: Changes in photosynthetic capacity and photosynthetic protein pattern during tomato fruit ripening. Plant Physiol. 1987, 84: 911-917. 10.1104/pp.84.3.911.
Barsan C, Sanchez-Bel P, Rombaldi C, Egea I, Rossignol M, Kuntz M, Zouine M, Latche A, Bouzayen M, Pech JC: Characteristics of the tomato chromoplast revealed by proteomic analysis. J Exp Bot. 2010, 61: 2413-2431. 10.1093/jxb/erq070.
Markovic O, Janecek S: Pectin degrading glycoside hydrolases of family 28: sequence-structural features, specificities and evolution. Protein Eng. 2001, 14: 615-631. 10.1093/protein/14.9.615.
Choudhury SR, Roy S, Sengupta DN: Characterization of cultivar differences in beta-1,3 glucanase gene expression, glucanase activity and fruit pulp softening rates during fruit ripening in three naturally occurring banana cultivars. Plant Cell Rep. 2009, 28: 1641-1653. 10.1007/s00299-009-0764-5.
Barka EA, Kalantari S, Makhlouf J, Arul J: Impact of UV-C irradiation on the cell wall-degrading enzymes during ripening of tomato (Lycopersicon esculentum L.) fruit. J Agric Food Chem. 2000, 48: 667-671. 10.1021/jf9906174.
Steele NM, Mccann MC, Roberts K: Pectin modification in cell walls of ripening tomatoes occurs in distinct domains. Plant Physiol. 1997, 114: 373-381.
Maclachlan G, Brady C: Endo-1,4-beta-glucanase, xyloglucanase, and xyloglucan endo-transglycosylase activities versus potential substrates in ripening tomatoes. Plant Physiol. 1994, 105: 965-974.
Saladie M, Rose JK, Cosgrove DJ, Catala C: Characterization of a new xyloglucan endotransglucosylase/hydrolase (XTH) from ripening tomato fruit and implications for the diverse modes of enzymic action. Plant J. 2006, 47: 282-295. 10.1111/j.1365-313X.2006.02784.x.
Miedes E, Lorences EP: Xyloglucan endotransglucosylase/hydrolases (XTHs) during tomato fruit growth and ripening. J Plant Physiol. 2009, 166: 489-498. 10.1016/j.jplph.2008.07.003.
Rose JKC, Lee HH, Bennett AB: Expression of a divergent expansin gene is fruit-specific and ripening-regulated. Proc Natl Acad Sci U S A. 1997, 94: 5955-5960. 10.1073/pnas.94.11.5955.
Rugkong A, Rose JK, Lee SJ, Giovannoni JJ, O’Neill MA, Watkins CB: Cell wall metabolism in cold-stored tomato fruit. Postharvest Biol Technol. 2010, 57: 106-113. 10.1016/j.postharvbio.2010.03.004.
Seymour GB, Chapman NH, Chew BL, Rose JK: Regulation of ripening and opportunities for control in tomato and other fruits. Plant Biotechnol J. 2013, 11: 269-278. 10.1111/j.1467-7652.2012.00738.x.
Tang GQ, Luscher M, Sturm A: Antisense repression of vacuolar and cell wall invertase in transgenic carrot alters early plant development and sucrose partitioning. Plant Cell. 1999, 11: 177-189.
Farrar J: Regulation of root weight ratio is mediated by sucrose: opinion. Plant Soil. 1996, 185: 13-19. 10.1007/BF02257561.
Godt DE, Roitsch T: Regulation and tissue-specific distribution of mRNAs for three extracellular invertase isoenzymes of tomato suggests an important function in establishing and maintaining sink metabolism. Plant Physiol. 1997, 115: 273-282. 10.1104/pp.115.1.273.
Gear ML, McPhillips ML, Patrick JW, McCurdy DW: Hexose transporters of tomato: molecular cloning, expression analysis and functional characterization. Plant Mol Biol. 2000, 44: 687-697. 10.1023/A:1026578506625.
McCurdy DW, Dibley S, Cahyanegara R, Martin A, Patrick JW: Functional characterization and RNAi-mediated suppression reveals roles for hexose transporters in sugar accumulation by tomato fruit. Mol Plant. 2010, 3: 1049-1063. 10.1093/mp/ssq050.
Brown MM, Hall JL, Ho LC: Sugar uptake by protoplasts isolated from tomato fruit tissues during various stages of fruit growth. Physiol Plant. 1997, 101: 533-539. 10.1111/j.1399-3054.1997.tb01034.x.
Sade D, Eybishtz A, Gorovits R, Sobol I, Czosnek H: A developmentally regulated lipocalin-like gene is overexpressed in Tomato yellow leaf curl virus-resistant tomato plants upon virus inoculation, and its silencing abolishes resistance. Plant Mol Biol. 2012, 80: 273-287. 10.1007/s11103-012-9946-6.
Alleva K, Marquez M, Villarreal N, Mut P, Bustamante C, Bellati J, Martinez G, Civello M, Amodeo G: Cloning, functional characterization, and co-expression studies of a novel aquaporin (FaPIP2;1) of strawberry fruit. J Exp Bot. 2010, 61: 3935-3945. 10.1093/jxb/erq210.
Hohnjec N, Vieweg ME, Puhler A, Becker A, Kuster H: Overlaps in the transcriptional profiles of Medicago truncatula roots inoculated with two different Glomus fungi provide insights into the genetic program activated during arbuscular mycorrhiza. Plant Physiol. 2005, 137: 1283-1301. 10.1104/pp.104.056572.
Liu J, Maldonado-Mendoza I, Lopez-Meyer M, Cheung F, Town CD, Harrison MJ: Arbuscular mycorrhizal symbiosis is accompanied by local and systemic alterations in gene expression and an increase in disease resistance in the shoots. Plant J. 2007, 50: 529-544. 10.1111/j.1365-313X.2007.03069.x.
Guether M, Balestrini R, Hannah M, He J, Udvardi MK, Bonfante P: Genome-wide reprogramming of regulatory networks, transport, cell wall and membrane biogenesis during arbuscular mycorrhizal symbiosis in Lotus japonicus. New Phytol. 2009, 182: 200-212. 10.1111/j.1469-8137.2008.02725.x.
Hogekamp C, Arndt D, Pereira PA, Becker JD, Hohnjec N, Kuester H: Laser microdissection unravels cell-type-specific transcription in arbuscular mycorrhizal roots, including CAAT-Box transcription factor gene expression correlating with fungal contact and spread. Plant Physiol. 2011, 157: 2023-2043. 10.1104/pp.111.186635.
Giovannetti M, Balestrini R, Volpe V, Guether M, Straub D, Costa A, Ludewig U, Bonfante P: Two putative-aquaporin genes are differentially expressed during arbuscular mycorrhizal symbiosis in Lotus japonicus. BMC Plant Biol. 2012, 12: 186-10.1186/1471-2229-12-186.
Barzana G, Aroca R, Paz JA, Chaumont F, Carmen Martinez-Ballesta M, Carvajal M, Manuel Ruiz-Lozano J: Arbuscular mycorrhizal symbiosis increases relative apoplastic water flow in roots of the host plant under both well-watered and drought stress conditions. Ann Bot. 2012, 109: 1009-1017. 10.1093/aob/mcs007.
Ruzicka D, Chamala S, Barrios-Masias FH, Martin F, Smith S, Jackson LE, Barbazuk WB, Schachtman DP: Inside arbuscular mycorrhizal roots - molecular probes to understand the symbiosis. Plant Genome. 2013, 6: 1-13.
Rausch C, Zimmermann P, Amrhein N, Bucher M: Expression analysis suggests novel roles for the plastidic phosphate transporter Pht2;1 in auto- and heterotrophic tissues in potato and Arabidopsis. Plant J. 2004, 39: 13-28. 10.1111/j.1365-313X.2004.02106.x.
Burleigh SH: Relative quantitative RT-PCR to study the expression of plant nutrient transporters in arbuscular mycorrhizas. Plant Sci. 2001, 160: 899-904. 10.1016/S0168-9452(00)00460-X.
Hildebrandt U, Schmelzer E, Bothe H: Expression of nitrate transporter genes in tomato colonized by an arbuscular mycorrhizal fungus. Physiol Plant. 2002, 115: 125-136. 10.1034/j.1399-3054.2002.1150115.x.
Herrera-Medina MJ, Steinkellner S, Vierheilig H, Bote JAO, Garrido JMG: Abscisic acid determines arbuscule development and functionality in the tomato arbuscular mycorrhiza. New Phytol. 2007, 175: 554-564. 10.1111/j.1469-8137.2007.02107.x.
Martin Rodriguez JA, Leon Morcillo R, Vierheilig H, Antonio Ocampo J, Ludwig-Mueller J, Garcia Garrido JM: Mycorrhization of the notabilis and sitiens tomato mutants in relation to abscisic acid and ethylene contents. J Plant Physiol. 2010, 167: 606-613. 10.1016/j.jplph.2009.11.014.
Foo E, Ross JJ, Jones WT, Reid JB: Plant hormones in arbuscular mycorrhizal symbioses: an emerging role for gibberellins. Ann Bot. 2013, 111: 769-779. 10.1093/aob/mct041.
Roxrud I, Lid SE, Fletcher JC, Schmidt ED, Opsahl-Sorteberg HG: GASA4, one of the 14-member Arahidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol. 2007, 48: 471-483. 10.1093/pcp/pcm016.
Burbidge A, Grieve T, Terry C, Corlett J, Thompson A, Taylor I: Structure and expression of a cDNA encoding zeaxanthin epoxidase, isolated from a wilt-related tomato (Lycopersicon esculentum Mill.) library. J Exp Bot. 1997, 48: 1749-1750.
Li Y, Walton DC: Violaxanthin is an abscisic-acid precursor in water-stressed dark-grown bean-leaves. Plant Physiol. 1990, 92: 551-559. 10.1104/pp.92.3.551.
Okamoto M, Kuwahara A, Seo M, Kushiro T, Asami T, Hirai N, Kamiya Y, Koshiba T, Nambara E: CYP707A1 and CYP707A2, which encode abscisic acid 8′-hydroxylases, are indispensable for proper control of seed dormancy and germination in Arabidopsis. Plant Physiol. 2006, 141: 97-107. 10.1104/pp.106.079475.
Zhang Z, Zhang H, Quan R, Wang XC, Huang R: Transcriptional regulation of the ethylene response factor LeERF2 in the expression of ethylene biosynthesis genes controls ethylene production in tomato and tobacco. Plant Physiol. 2009, 150: 365-377. 10.1104/pp.109.135830.
Lorenzo O, Piqueras R, Sanchez-Serrano JJ, Solano R: Ethylene response Factor1 integrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell. 2003, 15: 165-178. 10.1105/tpc.007468.
Kloppholz S, Kuhn H, Requena N: A secreted fungal effector of glomus intraradices promotes symbiotic biotrophy. Curr Biol. 2011, 21: 1204-1209. 10.1016/j.cub.2011.06.044.
Bleeker PM, Mirabella R, Diergaarde PJ, VanDoorn A, Tissier A, Kant MR, Prins M, de Vos M, Haring MA, Schuurink RC: Improved herbivore resistance in cultivated tomato with the sesquiterpene biosynthetic pathway from a wild relative. Proc Natl Acad Sci U S A. 2012, 109: 20124-20129. 10.1073/pnas.1208756109.
Ahuja I, Kissen R, Bones AM: Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17: 73-90. 10.1016/j.tplants.2011.11.002.
Walling LL: The myriad plant responses to herbivores. J Plant Growth Regul. 2000, 19: 195-216.
Falara V, Akhtar TA, Nguyen TT, Spyropoulou EA, Bleeker PM, Schauvinhold I, Matsuba Y, Bonini ME, Schilmiller AL, Last RL, Schuurink RC, Pichersky E: The tomato Terpene synthase gene family. Plant Physiol. 2011, 157: 770-789. 10.1104/pp.111.179648.
Kang JH, Shi F, Jones A, Marks M, Howe GA: Distortion of trichome morphology by the hairless mutation of tomato affects leaf surface chemistry. J Exp Bot. 2010, 61: 1053-1064. 10.1093/jxb/erp370.
Schilmiller AL, Schauvinhold I, Larson M, Xu R, Charbonneau AL, Schmidt A, Wilkerson C, Last RL, Pichersky E: Monoterpenes in the glandular trichomes of tomato are synthesized from a neryl diphosphate precursor rather than geranyl diphosphate. Proc Natl Acad Sci U S A. 2009, 106: 10865-10870. 10.1073/pnas.0904113106.
van Schie CC, Haring MA, Schuurink RC: Tomato linalool synthase is induced in trichomes by jasmonic acid. Plant Mol Biol. 2007, 64: 251-263. 10.1007/s11103-007-9149-8.
Morandi D: Occurrence of phytoalexins and phenolic compounds in endomycorrhizal interactions, and their potential role in biological control. Plant Soil. 1996, 185: 241-251. 10.1007/BF02257529.
Sundaresan P, Raja NU, Gunasekaran P: Induction and accumulation of phytoalexins in Cowpea roots infected with a mycorrhizal fungus Glomus-fasciculatum and their resistance to Fusarium-wilt disease. J Biosci. 1993, 18: 291-301. 10.1007/BF02703126.
Yao MK, Desilets H, Charles MT, Boulanger R, Tweddell RJ: Effect of mycorrhization on the accumulation of rishitin and solavetivone in potato plantlets challenged with Rhizoctonia solani. Mycorrhiza. 2003, 13: 333-336. 10.1007/s00572-003-0267-0.
Jones JD, Dangl JL: The plant immune system. Nature. 2006, 444: 323-329. 10.1038/nature05286.
Hao Z, Fayolle L, van Tuinen D, Chatagnier O, Li X, Gianinazzi S, Gianinazzi-Pearson V: Local and systemic mycorrhiza-induced protection against the ectoparasitic nematode **phinema index involves priming of defence gene responses in grapevine. J Exp Bot. 2012, 63: 3657-3672. 10.1093/jxb/ers046.
Campos-Soriano L, Garcia-Martinez J, San Segundo B: The arbuscular mycorrhizal symbiosis promotes the systemic induction of regulatory defence-related genes in rice leaves and confers resistance to pathogen infection. Mol Plant Pathol. 2012, 13: 579-592. 10.1111/j.1364-3703.2011.00773.x.
Li HY, Yang GD, Shu HR, Yang YT, Ye BX, Nishida I, Zheng CC: Colonization by the arbuscular mycorrhizal fungus Glomus versiforme induces a defense response against the root-knot nematode Meloidogyne incognita in the grapevine (Vitis amurensis Rupr.), which includes transcriptional activation of the class III chitinase gene VCH3. Plant Cell Physiol. 2006, 47: 154-163.
Zamioudis C, Pieterse CM: Modulation of host immunity by beneficial microbes. Mol Plant Microbe Interact. 2012, 25: 139-150. 10.1094/MPMI-06-11-0179.
Nagy R, Karandashov V, Chague W, Kalinkevich K, Tamasloukht M, Xu GH, Jakobsen I, Levy AA, Amrhein N, Bucher M: The characterization of novel mycorrhiza-specific phosphate transporters from Lycopersicon esculentum and Solanum tuberosum uncovers functional redundancy in symbiotic phosphate transport in solanaceous species. Plant J. 2005, 42: 236-250. 10.1111/j.1365-313X.2005.02364.x.
Czarnecki O, Yang J, Weston DJ, Tuskan GA, Chen JG: A dual role of strigolactones in phosphate acquisition and utilization in plants. Int J Mol Sci. 2013, 14: 7681-7701. 10.3390/ijms14047681.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M: MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37: 914-939. 10.1111/j.1365-313X.2004.02016.x.
Ouziad F, Wilde P, Schmelzer E, Hildebrandt U, Bothe H: Analysis of expression of aquaporins and Na+/H + transporters in tomato colonized by arbuscular mycorrhizal fungi and affected by salt stress. Environ Exp Bot. 2006, 57: 177-186. 10.1016/j.envexpbot.2005.05.011.
Jung SC, Martinez-Medina A, Lopez-Raez JA, Pozo MJ: Mycorrhiza-induced resistance and priming of plant defenses. J Chem Ecol. 2012, 38: 651-664. 10.1007/s10886-012-0134-6.
Pozo MJ, Cordier C, Dumas-Gaudot E, Gianinazzi S, Barea JM, Azcon-Aguilar C: Localized versus systemic effect of arbuscular mycorrhizal fungi on defence responses to Phytophthora infection in tomato plants. J Exp Bot. 2002, 53: 525-534. 10.1093/jexbot/53.368.525.
Hetherington SE, Smillie RM, Davies WJ: Photosynthetic activities of vegetative and fruiting tissues of tomato. J Exp Bot. 1998, 49: 1173-1181. 10.1093/jxb/49.324.1173.
Lytovchenko A, Eickmeier I, Pons C, Osorio S, Szecowka M, Lehmberg K, Arrivault S, Tohge T, Pineda B, Anton MT, Hedtke B, Lu Y, Fisahn J, Bock R, Stitt M, Grimm B, Granell A, Fernie AR: Tomato fruit photosynthesis is seemingly unimportant in primary metabolism and ripening but plays a considerable role in seed development. Plant Physiol. 2011, 157: 1650-1663. 10.1104/pp.111.186874.
Bravdo BA, Palgi A, Lurie S: Changing ribulose diphosphate carboxylase/oxygenase activity in ripening tomato fruit. Plant Physiol. 1977, 60: 309-312. 10.1104/pp.60.2.309.
Amaya-Carpio L, Davies F, Fox T, He C: Arbuscular mycorrhizal fungi and organic fertilizer influence photosynthesis, root phosphatase activity, nutrition, and growth of Ipomoea carnea ssp fistulosa. Photosynthetica. 2009, 47: 1-10. 10.1007/s11099-009-0003-x.
Black KG, Mitchell DT, Osborne BA: Effect of mycorrhizal-enhanced leaf phosphate status on carbon partitioning, translocation and photosynthesis in cucumber. Plant Cell Environ. 2000, 23: 797-809. 10.1046/j.1365-3040.2000.00598.x.
Mayzlish-Gati E, LekKala SP, Resnick N, Wininger S, Bhattacharya C, Lemcoff J, Kapulnik Y, Koltai H: Strigolactones are positive regulators of light-harvesting genes in tomato. J Exp Bot. 2010, 61: 3129-3136. 10.1093/jxb/erq138.
Dell′Amico J, Torrecillas A, Rodriguez P, Morte A, Sanchez-Blanco MJ: Responses of tomato plants associated with the arbuscular mycorrhizal fungus Glomus clarum during drought and recovery. J Agric Sci. 2002, 138: 387-393.
Boldt K, Poers Y, Haupt B, Bitterlich M, Kuehn C, Grimm B, Franken P: Photochemical processes, carbon assimilation and RNA accumulation of sucrose transporter genes in tomato arbuscular mycorrhiza. J Plant Physiol. 2011, 168: 1256-1263. 10.1016/j.jplph.2011.01.026.
Wright DP, Scholes JD, Read DJ: Effects of VA mycorrhizal colonization on photosynthesis and biomass production of Trifolium repens L. Plant Cell Environ. 1998, 21: 209-216. 10.1046/j.1365-3040.1998.00280.x.
Kaschuk G, Kuyper TW, Leffelaar PA, Hungria M, Giller KE: Are the rates of photosynthesis stimulated by the carbon sink strength of rhizobial and arbuscular mycorrhizal symbioses?. Soil Biol Biochem. 2009, 41: 1233-1244. 10.1016/j.soilbio.2009.03.005.
Crookes PR, Grierson D: Ultrastructure of tomato fruit ripening and the role of polygalacturonase isoenzymes in cell wall degradation. Plant Physiol. 1983, 72: 1088-1093. 10.1104/pp.72.4.1088.
Hewitt EJ: Sand and water culture methods used in the study of plant nutrition. 1966, England: Commonwealth Agricultural Bureaux
Trouvelot A, Kough JL, Gianinazzi-Pearson V: Mesure du taux de mycorhization VA d'un systeme radiculaire. Recherche de methodes d'estimation ayant une signification fonctionnelle. Physiological and Genetical Aspects of Mycorrhizae. Proceedings of the 1st European Symposium on mycorrhizae. Edited by: Gianinazzi-Pearson V, Gianinazzi S. 1984, Paris: Institut National de la Recherche Agronomique, 217-221.
Chang S, Puryear J, Cairney J: A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report. 1993, 11: 113-116. 10.1007/BF02670468.
Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17: 10-12.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7: 562-578. 10.1038/nprot.2012.016.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-10.1186/gb-2010-11-10-r106.
Tenore GC, Troisi J, Di Fiore R, Manfra M, Novellino E: Nutraceutical value and toxicological profile of selected red wines from Morocco. Food Chem. 2011, 129: 792-798. 10.1016/j.foodchem.2011.05.022.
Eberhardt MV, Lee CY, Liu RH: Nutrition - Antioxidant activity of fresh apples. Nature. 2000, 405: 903-904.
Singleton VL, Orthofer R, Lamuela-Raventos RM: Analysis of total phenols and other oxidation substrates and antioxidants by means of Folin-Ciocalteu reagent. Oxidants Antioxid A. 1999, 299: 152-178.
Nagata M, Yamashita I: Simple method for simultaneous determination of chlorophyll and carotenoids in tomato fruit. J Jpn Soc Food Sci Technol. 1992, 39: 925-928. 10.3136/nskkk1962.39.925.
Acknowledgements
We thank Linda Pastero and Emanuele Costa (Dipartimento Scienze della Terra), Stefania Daghino (DBIOS, University of Turin) and Paola Benzi (Department of Chemistry, University of Turin) for their support in the mineral nutrient content analysis, and Maria Teresa Della Beffa for maintaining the tomato cultures. This research was funded by the ArAs project (Regione Piemonte Poli d’Innovazione) and by the MYCOPLANT (Compagnia di San Paolo, Progetti di Ateneo) to PB. IZ and AS received fellowships from the Regione Piemonte and University of Torino.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IZ participated in the design of the experiment, carried out most of the experimental part, participated in the sequence analysis and drafted the manuscript. AS participated in the design of the experiment, sequence analysis, results interpretation and drafted the manuscript. MC performed the phenological observations and the biochemical experiments; LM participated in the sequence analysis, MN followed the plant cultures and the mycorrhization experiments and drafted the text; GCT coordinated the nutraceutical analyses; PBag performed the bioinformatics analysis and participated in results interpretation; PB conceived the study, participated in its design and coordination and wrote the manuscript. All authors read and approved the final manuscript.
Inès Zouari, Alessandra Salvioli contributed equally to this work.
Electronic supplementary material
12864_2013_7042_MOESM1_ESM.pdf
Additional file 1: Table S1: Mineral content (C, N, K, P, S) of fully ripe CONTROL and MYC tomato fruit. Values are means ± SD (n = 5). DW, dry weight. Data highlighted in bold are significantly different (Non parametric test Kruskal-Wallis p < 0.05). Table S2. Phenology and fruit yield of CONT and MYC plants: length of the vegetative period (expressed in days after transplanting, DAT), number of days from flowering to the first fruit, fruiting period length, average fruit number per plant, total fruit production, average fruit weight and total fruit yield. Data are expressed as mean ± standard deviation, n = 10. Statistically significant data are highlighted in bold according to the non-parametric Kruskal-Wallis test (p < 0.05). Table S4. Biochemical and nutraceutical parameters measured in fully ripe CONTROL and MYC tomato fruits. Data highlighted in bold are significantly different (one-way ANOVA with Tukey’s posthoc test, p < 0.05). Table S5. Genes and corresponding primers used in real-time RT-PCR. (PDF 83 KB)
12864_2013_7042_MOESM2_ESM.pdf
Additional file 2: Figure S1: Mycorrhization parameters of tomato roots inoculated with the AM fungus Funneliformis mosseae. F%: colonization frequency; M%: mycorrhizal colonization intensity; A%: arbuscule abundance; a%: arbuscule abundance in the mycorrhized portion. Mycorrhization parameters were determined according to Trouvelot method (1986). Values are means ± SD (n=5). For each plant, 100 cm of root were measured. Figure S2. Classification of tomato fruit expressed genes. All expressed genes (cutoff RPKM>0.1) are represented. The number of transcripts assigned to tomato biological process (GOSLIM plant map**) is shown in parentheses. Figure S3. Transcriptional changes induced by mycorrhization in tomato fruits. Normalized expression mean values are plotted versus log2 fold changes and called DEGs (FDR<0.1) are plotted in red. Blue horizontal lines indicate a two-fold change in expression values for up- and down-regulated genes. Figure S4. Sesquiterpenoid phytoalexin biosynthesis pathway. Figure S5. q-PCR data for five selected genes involved in carotenoid metabolism on fruits from mycorrhizal and control plants. The y axis represents ∆CT value (Ct gene-Ct ubiquitin). Data are mean ± SD for n=3. Identities of genes are: Lox (lipoxygenase TomloxC); Hdr (hydroxymethylbutenyl diphoshate reductase); Dxs (deoxyxylulose 5-phosphate synthase); Psy (phytoene synthase); Ccd (carotenoid cleavage dioxygenase). Figure S6. Statistical analyses of q-PCR data for nine differentially regulated genes in three ripening stages green (G), breaker (B) and turning (T). The y axis represents ∆CT value (Ct gene-Ct ubiquitin). Data are mean ± SD for n = 3. One-way ANOVA (Tukey’s posthoc test) was used for data analysis and asterisks indicate statistical significance at p<0.05. Identities are as follows: Solyc01g008540.2.1 (Cinnamoyl CoA reductase-like protein); Solyc02g078400.2 (Allantoinase); Solyc02g076920.2.1 (Transcription factor MYC2); Solyc10g005080.2.1 (Late elongated hypocotyl and circadian clock associated-1-like); Solyc09g066410.1.1 (Inorganic phosphate transporter); Solyc06g062540.2.1 (Putative acid phosphatase); Solyc03g083910.2.1 (Acid beta-fructofuranosidase); Solyc01g080280.2.1 (Glutamine synthetase); Solyc10g055390.1 (Nodulin family protein). (PDF 1 MB)
12864_2013_7042_MOESM3_ESM.xlsx
Additional file 3: Table S3: Genes identified as differentially regulated in MYC fruit in comparison with CONT fruit. Genes are ranked in descending order of their log2 fold change value. (XLSX 81 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zouari, I., Salvioli, A., Chialva, M. et al. From root to fruit: RNA-Seq analysis shows that arbuscular mycorrhizal symbiosis may affect tomato fruit metabolism. BMC Genomics 15, 221 (2014). https://doi.org/10.1186/1471-2164-15-221
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-221