
Abstract
The influence of a pancreatic enzyme, lipase, on doxorubicin-loaded anionic liposomes, their complexes with spherical polycationic brushes, and ternary complexes with brushes and bovine serum albumin has been studied. The two-stage kinetics of the enzyme-induced disturbance of liposomal membrane integrity has been investigated using fluorescence spectroscopy. A 120-min induction period free of any serious damage to liposome membranes has been found for all enzyme-treated samples. This period is followed by rapid release of encapsulated doxorubicin. Enzymatic hydrolysis of the liposomes and their binary complexes with brushes has been found to proceed at the same rate, while a rather long resistance to the disturbance of liposome integrity has been observed for the ternary complexes. It has been assumed that albumin plays the role of a sterically screening agent, which hinders the enzymatic hydrolysis in multiliposomal complexes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Encapsulation of drugs is one of the most popular strategies to increase their efficiency. Different types of carriers, such as polymers, micelles, cyclodextrins, and emulsions, can be used to enhance the solubility and circulation time of drugs, protect them from hydrolysis, etc. [1–4]. Liposomes are biocompatible nanocontainers that are capable of encapsulating hydrophilic substances in the inner water cavities of bilayer lipid vesicles [5]. Liposomes may be used as individual nanocarriers and components of complex multiliposomal systems [6, 7]. Since liposomes consist of lipids, they are prone to enzymatic hydrolysis with lipases presented in organism [8]. As a rule, lipases are located in the digestive system of human body [9]; however, high concentrations of lipases have been registered in some inflammation regions. For example, phospholipase A2 has been shown to promote inflammation in mammals, thus facilitating the formation of fatty acids, including arachidonic acid. Arachidonic acid is then metabolized to form several inflammatory and thrombogenic agents. Excess levels of phospholipase A2 are thought to contribute to several inflammatory diseases and have been shown to promote vascular inflammation, which correlates with coronary events accompanying coronary artery disease and acute coronary syndrome [10]. Thus, the liposomal containers specially intended for anti-cancer treatment may undergo undesired disruptive action of the enzymes, which hydrolyze ester bonds in lipids, thereby disturbing the integrity of liposomal membranes and resulting in undesired release of substances encapsulated in the nanocontainers.
Complexation of the liposomes with polyelectrolytes is a promising strategy for the protection of lipids against lipase action [11]. Polyelectrolytes are supposed to form barrier layers between lipids and enzymes. The efficiency of the steric isolation of liposomal membranes from hydrolyzing enzymes with the use of macromolecules was mentioned in [12] by J. Han et al. However, the authors of [13] have shown that the modification of a didecanoylglycerol lipid monolayer with chitosan does not protect the surface from pancreatic lipase. Moreover, polyelectrolyte–liposome complexes are characterized by diverse spatial arrangements of macromolecules and vesicles depending on the nature of polymers, their architecture, sizes, etc. [14–18]. Hence, a deterioration or the complete loss of the protective effect of a polymer with respect to enzymatic hydrolysis may be expected for the structured “core–shell” complexes, whose liposomes form an external layer around the polyelectrolyte–core particles.
The goal of this work is to analyze the behavior of multiliposomal containers in biological fluids; the action of a hydrolytic enzyme, lipase, on the liposomes included into anionic liposome–spherical polycationic brush (SPB) complexes; and the feasibility of the practical application of these containers.
EXPERIMENTAL
Materials
SPBs were supplied by professor Ballauff. They were 85-nm polystyrene particles containing grafted cationic poly(aminoethyl methacrylate) hydrochloride chains with average degree of polymerization n equal to 695 and an average interchain distance of 5.8 nm [19]; thus, average number N of the chains per brush was 675. Concentration of SPBs [SPB+] was determined by titration with a sodium polystyrenesulphonate solution and expressed in moles of protonated amino groups per liter.
Electroneutral lipids, dioleoylphosphatidylcholine (DOPC) and palmitoyloleoylphosphatidylserine (POPS) produced by Avanti Polar Lipids Co. were used in the work. In addition, bovine serum albumin (BSA), Lipase II extracted from porcine pancreas (Serva), and doxorubicin hydrochloride (Dox) (Teva) were used in the experiments.
The structures of lipids, Dox, and SPBs are shown in Fig. 1.

Structures of the used reagents.
Small unilamellar liposomes were prepared by sonication [20]. Weighted portions of DOPC and POPS were dissolved in a methanol/chloroform mixture in a manner such that the molar fraction of [POPS]/([DOPC] + [POPS]) anionic lipid was 0.3. Then, the organic solvent was evaporated in a Laborota vacuum rotary evaporator. A resulting thin lipid film was dispersed in 0.01 M Tris/NaCl buffer with pH 7.0 and a NaCl concentration of 0.15 M. The freshly formed suspension was sonicated with a Cole-Parmer CP-750 instrument during two 5-min cycles. The titanium tip dust was separated from the prepared liposomes by centrifugation in an Eppendorf Mini-spin centrifuge at 10 000 rpm for 5 min. The average size of the liposomes was within a range of 40–60 nm. The average electrophoretic mobility (EPM) of the liposomes was –3.2 ± 0.3 (μm/s)/(V/cm). The samples were used within 24 h after the preparation.
Small unilamellar POPS/DOPC liposomes, which had anionic lipid molar fraction ν = [POPS]/([POPS] + [DOPC]) = 0.3 and contained fluorescent dye, doxorubicin, were prepared by the same procedure, while dispersing the lipid film in an acetate buffer with pH 4 [20]. The obtained liposomes were passed through a Sepharose CL4B column equilibrated with a Tris buffer (pH 7) to replace the extraliposomal solution. The addition of a Dox solution to the liposome dispersion was accompanied by the development of a pH gradient, which caused dye encapsulation. The process was monitored by fluorescence spectroscopy. The loading efficiency was 99%.
The Tris buffer containing 0.15 M NaCl was used in all experiments.
Investigation Methods
Hydrodynamic particle diameters in the suspension were determined by dynamic light scattering at a fixed angle of 90° with a Brookhaven Zeta Plus instrument equipped with a temperature-controlled cell.
Fluorescence intensity in a suspension of fluorescence-labeled liposomes was measured with an F‑4000 fluorimeter (Hitachi) operating at λem = 591 nm (λex = 530 nm).
BSA concentration was measured with a Hitachi UV-mini 1240 spectrophotometer at a wavelength of 280 nm, taking the extinction coefficient equal to 40 700 mol/(cm L), as determined from a calibration line.
Unbound BSA was separated from the complexes by centrifuging in an Eppendorf 5430 centrifuge (15 000 rpm, 10 min) using centrifuge tubes having filtering bottoms with a pore size of 100 nm.
RESULTS AND DISCUSSION
Saturated SPB–liposome binary complexes were obtained by direct mixing the suspensions of SPBs and liposomes in Tris/NaCl buffer with pH 7 as described in [21]. For the suspension of brushes with [SPB+] = 0.00014 base-mol/L, the concentration of liposomes was 0.65 mg/mL.
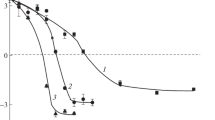
The influence of the enzyme on the integrity of liposomes was studied by fluorescence spectroscopy. First, lipase-induced hydrolysis was studied for individual liposomes. The dependences of the relative fluorescence of liposomes containing incorporated fluorescent dye, Dox, on incubation time are presented in Fig. 2.

Time dependences of Dox fluorescence intensity in (1, 2) suspensions of liposomes and (3, 4) SPB–liposome binary systems in the (1, 3) absence and (2, 4) presence of lipase. Polycation concentration is [SPB+] = 1.4 × 10–4 base-mol/L, liposome concentration is 0.65 mg/mL, and lipase concentration is 0.1 mg/mL.
For suspensions of liposomes containing Dox in the self-quenching concentration, the relative fluorescence intensity remained unchanged for, at least, 4 h (Fig. 2, curve 1). This indicated that the lipid membrane remained integral [22]. The addition of type-II porcine pancreatic lipase to a suspension of liposomes induced a two-stage process represented by curve 2 in Fig. 2. The fluorescence remained unchanged for 120 min after lipase was added. Then the fluorescence rapidly increased, thus indicating a damage of the liposomes and the release of the fluorescent dye into the ambient solution. The complete release of the Dox was detected after 260-min incubation. The induction periods seen in curves 2 and 4 may be related to the time required for the enzyme to interact with the lipid membrane and form the enzyme–lipid-containing substrate complex prone to the hydrolysis. This process is especially pronounced for liposomes, whose surface is, to a substantial extent, modified with silicon nanoparticles [23]. However, in some works, it was reported that, although the products of lipid hydrolysis, i.e., fatty acids, began to be accumulated immediately after the enzyme was added, partial hydrolysis did not give rise to the formation of membrane defects able to provide the dye release. Therewith, the induction period could last from a few seconds to several hours [24]. The duration of the induction period preceding the appearance of liposomal membrane defects that promote the release of the encapsulated compound is primarily determined by the nature of lipids, i.e., the length of “tails,” the shape and packing density of their molecules, and the presence of lipid domains and defective regions in lipid monolayers [25]. At the same time, when studying liposomes with a modified surface, the authors of [23] observed an induction period of the enzymatic hydrolysis during which no hydrolysis products were accumulated. Rades et al. reported long-term hydrolysis of liposomes that did not break the bilayer [26]. For 60 min, liposomes composed of soy-bean lecithin or hydrated egg lecithin retained their sizes, while the hydrolysis products started to be accumulated just after the onset of the lipolysis. Mouritsen et al. [24] have assumed that the enzyme-induced acceleration of the release of hydrophilic substances encapsulated in liposomes, which occurs after the induction period, is triggered by the accumulation of the hydrolysis products and the formation of defects in the membranes as a result of the initial action of the enzyme. At present, there is no detailed review devoted to the lipase-mediated hydrolysis of small unilamellar mixed liposomes. It is usually noted that the destabilization of the bilayer begins as soon as lipase and liposomes start interacting [27, 28]. Then, this process leads to the avalanche-like growth of defects formed in the bilayer. However, cases were reported in which the hydrolysis did take place in the outer monolayers of liposomes but did not destroy the liposomes [29].
The lipase sample had a negative total EPM value of –1.1 ± 0.1 (μm/s)/(V/cm). Hence, we could expect competitive binding of lipase and liposomes to the cationic chains of SPBs. The average hydrodynamic diameter of lipase was 65 nm. Therefore, taking into account the nonspherical shape of enzyme molecules, it could penetrate into the external layer of the polycationic chains of the brushes.
In the absence of lipase, saturated SPB–liposome binary complexes retained their integrity for at least 260 min (Fig. 2, curve 3). When the enzyme was added to the suspension of the binary complexes, the time dependence of the Dox release was similar to the time dependence of this fluorophore release for the unbound liposome/lipase mixture (Fig. 2 curve 4). The induction period was found to be 120 min, and the dye was completely released after 260-min incubation. Thus, the Coulomb interaction between SPBs and lipase in the Tris/NaCl buffer is insufficient, and the lipase–lipid membrane interaction is more preferable for the system. The binary complex has the core-shell structure, where the “core” is represented by an SPB, while the “shell” is formed by a layer of liposomes. Thus, only the “bottom” of the liposomes carries lipids that are capable of the direct interaction with polycationic chains of the SPBs. In turn, the grafted polycationic chains cannot cover the large area of the liposomes, but rather form only irregular salt bonds. This makes the majority the liposome surface to be accessible for the enzyme attack. Hence, the same profile of the kinetic curves for the hydrolysis of free liposomes and liposomes contained in the binary complexes shows that the complexation with SPBs does not impart an additional stability to liposomes with respect to enzymes. The authors of [14] observed a similar effect when studying the hydrolysis rate of the monolayers of diacylglycerol and its complexes with chitosan.
It may be assumed that the density of the grafted chains at the places of the contact with liposomes is very low; therefore, they do not “protect” the liposomes from the attacks of lipase. Taking into account that the hydrodynamic diameter of a brush complexed with liposomes in the Tris/NaCl buffer is ~200 nm, it can be shown that the distance between the ends of the grafted chains of SPBs is 5–7 times larger than the distance between the chain bases. On the other hand, the surface area of the liposomes brought in contact with the polycationic chains amounts to almost a half of the total surface area of the vesicles [30]. Hence, the filling of the excluded volume of the grafted chains of SPBs with dense particles would be able to partly “protect” liposomes from the action of lipase. Thus, the access of lipase to the surface of complexed liposomes can be hindered by modifying the SPB–liposome binary complex with densely packed molecules of a protein, such as albumin.
The isoelectric point of BSA is pI = 4.6 [31]. In the Tris buffer with рН 7.0, the EPM of protein globules is 1.2 ± 0.6 (μm/s)/(V/cm), which allows them to be adsorbed on the cationic brush surface. A BSA molecule has the shape of a prolate ellipsoid with sizes of 140 × 40 × 40 Å3 [32]; therefore, it can penetrate deep into brush coronas. The bonding of BSA with SPB was quantitatively estimated using the following procedure. Several mixtures were prepared with a constant SPB concentration and different concentrations of BSA. In 30 min, the suspensions were centrifuged in tubes equipped with 100-nm filters for 10 min at 15 000 rpm in an Eppendorf 5430 centrifuge. Such centrifugation was sufficient to separate all unbound BSA from the complexes. The UV spectra of the transparent supernatants were measured, and the optical densities were recalculated to BSA concentrations (Fig. 3) using a calibration plot.

Dependences of BSA concentration in supernatants of (1) SPB–albumin complexes and (2) SPB–liposome–albumin complexes on BSA concentration in the systems. (1, 2) Polycation concentration is [SPB+] = 1.4 × 10–4 base-mol/L and (2) liposome concentration is 0.65 mg/mL.
As follows from curve 1 in Fig. 3, the presence of BSA in the supernatants was not detected up to its concentration in the system of 0.22 mg/mL. Then, the concentration of BSA in the supernatant increased linearly with its concentration in the mixture with SPBs. The hydrodynamic diameter measured for the saturated BSA–SPB complex was 220 ± 10 nm. To analyze the corona volume occupied by BSA, let us perform the following calculations. In the Tris/NaCl buffer, the polycationic chains are unfolded and nonoverlapped [33]. The influence of counterions can be ignored, because the interaction of polycations with BSA macromolecules is accompanied by the release of the counterions into an ambient solution [34, 35]. Excluded volume Ve of the brushes, i.e. the particle volume unoccupied by poly(aminoethyl methacrylate) monomer units, was calculated as
where Vm is the volume of a poly(aminoethyl methacrylate) monomer unit [36], n is the degree of chain polymerization, N is the number of the chains per SPB, and Vc is the volume of SPB corona calculated as
where RSPB is the radius of an SPB and RPS is radius of the polystyrene core of the SPB.
These calculations have led us to conclude that less than 2% of the corona volume is occupied by aminoethyl methacrylate.
Fraction S of the corona volume occupied by BSA was calculated as follows:
where \(C_{{{\text{BSA}}}}^{*}\) is albumin concentration in saturated complex and NA is Avogadro’s number. It was found that S = 65%. Guo et al. have revealed that, in a mixture with cationic SPBs, BSA does not form bridging salt bonds between paired brushes and fills the corona volume until saturation is reached [37]. The authors of [37] have assumed that BSA molecules are nonuniformly distributed inside the corona, and their content is increased near the polystyrene core. The accommodation density of the SPB corona with BSA molecules decreases with an increase in the distance from the core.
Saturated SPB–liposome–albumin ternary complexes were obtained by directly mixing suspensions of saturated SPB–liposome binary complexes and BSA in the Tris/NaCl buffer with pH 7 as described in [21]. The bonding of BSA with the SPB–liposome complex was evaluated by the procedure described above. Several mixtures were prepared with the same SPB–liposome complex concentration and different concentrations of BSA. Thirty minutes later, the suspensions were centrifuged in tubes having filtering bottoms with a pore diameter of 100 nm for 10 min at 15 000 rpm in an Eppendorf 5430 centrifuge. Such centrifugation was sufficient to separate all unbound BSA from the complexes. The UV spectra of the transparent supernatants were then measured and the optical density values were recalculated to BSA concentrations (Fig. 3) using a calibration plot.
As follows from curve 2 in Fig. 3, BSA was absent in the supernatants up to a protein concentration in the system of 0.11 mg/mL. Then, the BSA concentration in the supernatant increased linearly with its concentration in the mixture with SPBs. The hydrodynamic diameter measured for the saturated SPB–liposome–albumin ternary complex was 240 ± 10 nm.
The physico-chemical aspects of the formation of saturated SPB–liposome–albumin ternary complexes were described previously [21]. Briefly, in ternary complexes, BSA is located in the corona of grafted chains of SPBs. Therewith, BSA molecules do not rupture the salt bounds between anionic lipids and cationic chains of SPBs. The fraction of the excluded volume occupied by BSA in the ternary complex was calculated by Eq. (3). This fraction has appeared to be 33%. It is reasonable to assume that BSA is relatively uniformly distributed over the polycationic corona of an SPB with the exception of the volume occupied by liposomes. Let us consider the influence of the satirical screening on the enzymatic hydrolysis of liposomes complexed with SPBs “decorated” with BSA.
The integrity of the liposomes in the saturated SPB– liposome–albumin ternary complex was controlled by fluorescence spectroscopy. No release of the dye from the Dox-filled liposomes included into the in ternary complex was detected for 260 min of incubation (Fig. 4 curve 1). The profile of dye release from the ternary complex in the presence of lipase is presented in Fig. 4 by curve 2. During the first 120 min of incubation, no release of Dox into the ambient medium was detected. Then, the fluorescence, which reflected the disturbance of the membrane integrity, increased. However the rate of fluorophore release was lower than that in the suspensions of SPB–liposome binary complexes. After 260-min incubation, less than 45% of Dox was released. Hence, the modification of the multiliposomal complex with albumin had increased the resistance of the complex to the action of lipase. The partial steric screening of the liposomes significantly prolong the lifetime of the multiliposomal nanocontainers.

Time dependences of Dox fluorescence in suspensions of SPB–liposome–albumin ternary complex in the (1) absence and (2) presence of lipase. Polycation concentration is [SPB+] = 1.4 × 10–4 base-mol/L, liposome concentration is 0.65 mg/mL, and lipase concentration is 0.1 mg/mL.
An additional possible scenario of the BSA-induced stabilization of the integrity of liposomes included into the ternary complexes should be discussed. As has been mentioned above, the drastic (avalanche-like) release of Dox from the liposomes after the induction period in the presence of the enzyme can be related to a dramatic change in the membrane composition due to the accumulation of hydrolysis products [24]. The products of the lipase-catalyzed hydrolysis are fatty acids, which, even being present in small amounts in a membrane, destabilize the liposomal bilayers in complexes with polycations due to a change in the membrane curvature [38]. In turn, a BSA molecule contains regions with a high affinity for fatty acids and is capable of binding with them [39]. Thus, the protein–fatty acid interaction may take place and stabilize the liposomal membrane. However, this assumption requires additional investigations.
Activity of lipases is one of the most important processes governing the lifetime of liposomal nanocontainers, including multiliposomal complexes, in human body. Enzymatic hydrolysis disturbs the integrity of liposomes, thereby decreasing the functional efficiency of the nanocontainers. The formation of SPB–anionic liposome complexes has no effect on the lipase-induced hydrolysis of the liposomes to compare with free ones. The total negative charge of the enzyme does not ensure electrostatic immobilization (which can deactivate the enzyme) of lipase on the brushes. Moreover, in spite of the relatively large diameter of a lipase molecule, the partial steric screening of liposomes by SPBs does not hinder the enzymatic hydrolysis, which occurs at a rate comparable with the hydrolysis rate of free liposomes. In turn, the modification of multiliposomal complexes with albumin decelerates the hydrolysis, thus protecting the nanocontainers from the destructive action of pancreatic lipase. This effect can be explained by the appearance of marked steric hindrances for lipase molecules approaching liposome surface, which, in turn, appears to be unprotected in the case of free vesicles and vesicles included into the binary complexes.
REFERENCES
Izumrudov, V.A., Mussabayeva, B.Kh., Kassymova, Zh.S., Klivenko, A.N., and Orazzhanova, L.K., Russ. Chem. Rev., 2019, vol. 88, p. 1046.
Torchilin, V.P., Curr. Drug Delivery, 2005, vol. 2, p. 319.
Spiridonov, V.V., Panova, I.G., Topchieva, I.N., and Zakharov, A.N., RSC Adv., 2016, vol. 6, p. 44113.
Singh, Y., Meher, J.G., Raval, K., Khan, F.A., Chaurasia, M., Jain, N.K., and Chourasia, M.K., J. Control Release, 2017, vol. 252, p. 28.
Zakharova, L.Ya., Kashapov, R.R., Pashirova, T.N., Mirgorodskaya, A.B., and Sinyashin, O.G., Mendeleev Commun., 2016, vol. 26, p. 457.
Tretiakova, D.S., Khaidukov, S.V., Babayants, A.A., Frolova, I.S., Shcheglovitova, O.N., Onishchenko, N.R., and Vodovozova, E.L., Acta Nat., 2020, vol. 12, p. 99.
Yaroslavov, A.A., Efimova, A.A., Sybachin, A.V., Chvalun, S.N., Kulebyakina, A.I., and Kozlova, E.V., RSC Adv., 2015, vol. 5, p. 31460.
Lasic, D.D., Trends Biotechnol., 1998, vol. 16, p. 307.
Armand, M., Curr. Opin. Clin. Nutr. Metab. Care, 2007, vol. 10, p. 156.
Mallat, Z., Lambeau, G., and Tedgui, A., Circulation, 2010, vol. 122, p. 2183.
Jain, P., Jain, S., Prasad, K.N., Jain, S.K., and Vyas, S.P., Mol. Pharm., 2009, vol. 593, p. 6.
Liu, W., Ye, A., Han, F., and Han, J., Adv. Colloid Interface Sci., 2019, vol. 263, p. 52.
Souza, A.L., Pavinatto, F.J., Caseli, L., Volpati, D., Miranda, P.B., and Oliveira, O.N., Colloids Surf. B, 2014, vol. 123, p. 870.
Ivashkov, O.V., Sybachin, A.V., Efimova, A.A., Pergushov, D.V., Orlov, V.N., Schmalz, H., and Yaroslavov, A.A., ChemPhysChem, 2015, vol. 16, p. 2849.
Wang, W.X., Gao, J.Q., and Liang, W.Q., Drug Delivery, 2011, vol. 18, p. 208.
Billard, A., Pourchet, L., Malaise, S., Alcouffe, P., Montembault, A., and Ladaviere, C., Carbohydr. Res., 2015, vol. 115, p. 651.
Yaroslavov, A.A., Sybachin, A.V., Zaborova, O.V., Pergushov, D.V., Zezin, A.B., Melik-Nubarov, N.S., Plamper, F.A., Müller, A.H.E., and Menger, F.M., Macromol. Biosci., 2014, vol. 14, p. 491.
Yaroslavov, A.A., Efimova, A.A., Sybachin, A.V., Izumrudov, V.A., Samoshin, V.V., Potemkin, I.I., Colloid J., 2011, vol. 73, p. 430.
Guo, X. and Zhao, K., J. Phys. Condens. Matter, 2017, vol. 29, p. 295101.
Yaroslavov, A.A., Panova, I.G., Sybachin, A.V., Spiridonov, V.V., Zezin, A.B., Mergel, O., Gelissen, A., Tiwari, R., Plamper, F.A., Richtering, W., and Menger, F.M., Nanomedicine, 2017, vol. 13, p. 1491.
Sybachin, A.V., Stepanova, D.A., Melik-Nubarov, N.S., and Yaroslavov, A.A., Polym. Sci. Ser. A, 2020, vol. 62, p. 32.
Le-Deygen, I.M. and Kudryashova, E.V., Moscow Univ. Chem. Bull., 2018, vol. 73, p. 69.
Mohanraj, V.J., Barnes, T.J., and Prestidge, C.A., Int. J. Pharm., 2010, vol. 392, p. 285.
Arouri, A., Hansen, A.H., Rasmussen, T.E., and Mouritsen, O.G., Curr. Opin. Colloid Interface Sci., 2013, vol. 18, p. 419.
Honger, T., Jorgensen, K., Biltonen, R.L., and Mouritsen, O.G., Biochemistry, 1996, vol. 35, p. 9003.
Parmentier, J., Thomas, N., Mullertz, A., Fricker, G., and Rades, T., Int. J. Pharm., 2012, vol. 437, p. 253.
Cheong, I. and Zhou, S., Methods Enzymol., 2009, vol. 465, p. 251.
Müller, R.H., Rühl, D., and Runge, S.A., Int. J. Pharm., 1996, vol. 144, p. 115.
Davies, D.E., Critchlow, M., Grainger, D.W., Reichert, A., Ringsdorf, H., and Lloyd, J.B., Biochim. Biophys. Acta, 1991, vol. 1084, p. 29.
Yaroslavov, A.A., Sybachin, A.V., Zaborova, O.V., Orlov, V.N., Ballauff, M., Talmon, Y., and Menger, F.M., Chem.-Eur. J., 2013, vol. 19, p. 13674.
Generalova, A.N. and Zubov, V.P., Polym. Sci. Ser. B, vol. 58, p. 385.
Wright, A.K. and Thompson, M.R., Biophys. J., 1975, vol. 15, p. 137.
Zhulina, E.B. and Borisov, O.V., Langmuir, 2011, vol. 27, p. 10615.
Zezin, A.B., Mikheikin, S.V., Rogacheva, V.B., Zansokhova, M.F., Sybachin, A.V., and Yaroslavov, A.A., Adv. Colloid Interface Sci., 2015, vol. 226, p. 17.
Kayitmazer, A.B., Seeman, D., Minsky, B.B., Dubin, P.L., and Xu, Y., Soft Matter, 2013, vol. 9, p. 2553.
https://www.polymerdatabase.com/polymers/Poly2-hydroxypropylmethacrylate.html.
Wang, W., Li, L., Henzler, K., Lu, Y., Wang, J., Han, H., Tian, Y., Wang, Y., Zhou, Z., Lotze, G., Narayanan, T., Ballauff, M., and Guo, X., Biomacromolecules, 2011, vol. 18, p. 1574.
Efimova, A.A., Sybachin, A.V., and Yaroslavov, A.A., Polym. Sci. Ser. C, 2011, vol. 53, p. 18.
Gew, L.T. and Misran, M., Nanoscale Res. Lett., 2014, vol. 9, p. 218.
Funding
The work was supported by the Russian Foundation for Basic Research (project no. 19-03-00314).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of int-erest.
Additional information
Translated by A. Kirilin
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sybachin, A.V., Stepanova, D.A. Modification of Multiliposomal Nanocontainers with Albumin as a Method for Increasing Their Resistance to Enzymatic Hydrolysis. Colloid J 83, 252–258 (2021). https://doi.org/10.1134/S1061933X21020113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061933X21020113